Анализ качества чтений
Команда: fastqc chr10.1.fastq
Результат:
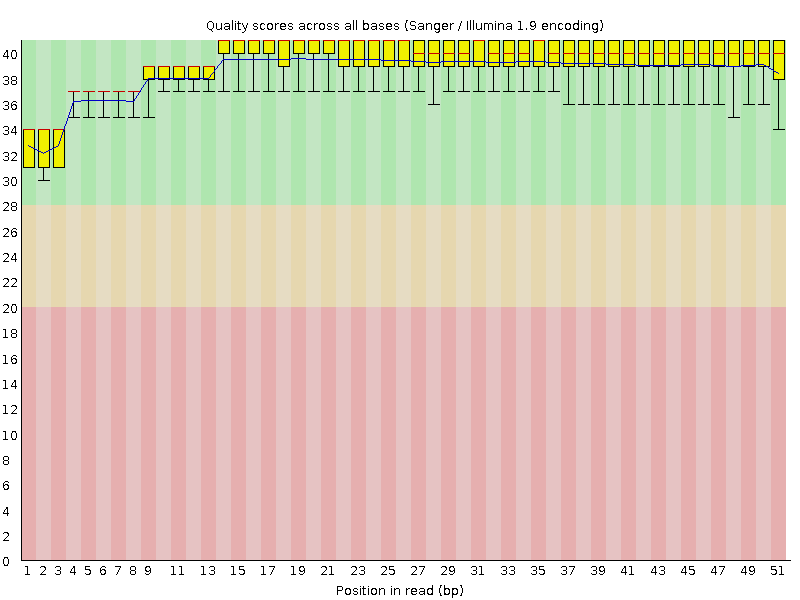
Результат:
Картирование чтений
| Картирование: | hisat2-build chr10.fasta chr10 |
| Выравнивание: | hisat2 --no-softclip -x chr10 -U chr10.1.fastq -S chr10_ali.sam |
По сравнению с предыдущем выпавниванием, был убран параметр,
запрещающий разрывное выравнивание, т. к. на этот раз мы работаем с РНК
в которой имеют место перегруппировки.
Анализ выравнивания
| Перевод в бинарный формат: | samtools view chr10_ali.sam -b -o chr10_ali.bam |
| Сортировка: | samtools sort -T /tmp/chr10_sorted -o chr10_sorted.bam chr10_ali.bam |
| Индексирование: | samtools index chr10_sorted.bam |
| Информация о картировании: | samtools idxstats chr10_sorted.bam |
В результате оказалось 15257 картированых ридов и 205 некартированых.
Подсчет чтений
htseq-count -f bam -i gene_id -s no -m union chr10_sorted.bam gencode.v19.chr_patch_hapl_scaff.annotation.gtf > count.txt
Опции:
Параметр -f выставляем bam. -s нам необходим no. -i в нашем случае gene_id. -m я решил попробовать union: Таким образом попадание в ген будет засчитываться, даже если часть рида в него легла.
Опции:
- -f - Формат: bam, sam
- -s - Цепь: yes(прямая), no(обе), reverse(обратная)
- -i - Атрибут GFF
- -m - Режим: union, intersection-strict, intersection-nonempty
Параметр -f выставляем bam. -s нам необходим no. -i в нашем случае gene_id. -m я решил попробовать union: Таким образом попадание в ген будет засчитываться, даже если часть рида в него легла.
Анализ результатов
- ENSG00000165732.8 - 14643
- ENSG00000266122.1 - 3
- no_feature - 611
- ambiguous - 0
- too_low_aQual - 0
- not_aligned - 205
- alignment_not_unique - 0
14643 ридов легли в ген DDX21 или DExD-box helicase 21. Он кодирует белок Nucleolar RNA helicase 2.
3 рида легли в ген RN7SL373P или RNA, 7SL, cytoplasmic 373, pseudogene. Он кодирует некодирующую РНК.
Ещё 611, видимо легли в некодирующие участки, а 205 были некартированы.