Практикум 8. Сигналы и мотивы
1. Описание мотива в белках паттерном
Для анализа семейства белков бактерий был выбран белок MreB - Murein region B (отвечает за поддержание формы клетки).
Сначала выровняли последовательности 8 белков с такой мнемоникой для разных бактерий.
Идентификаторы выбранных белков: MREB_BACSU, MREB_ECOLI, MREB_HAEIN, MREB_CAUVN, MREB_SALTY, MREB_PASMU, MREB_SHIFL, MREB_SALTI
Ссылка на выравниваниеПаттерн для консервативного участка в выравнивании без гэпов длиной 15 позиций: G-S-M-V-[VI]-D-I-G-G-G-T-T-E-V-A. Номера позиций в выравнивании: 162-176. Номера остатков в MREB_ECOLI: 160-174.
Далее по этому паттерну с помощью программы fuzzpro проводился поиск среди всех белков бактерий из Swiss-Prot.
Теперь определим точность поиска (число правильно найденных - TP, ложноположительных находок - FP и ложноотрицательных результатов - FN). Число TP - можно определить, проанализировав мнемоники найденных последовательностей в файле mreb_search.fuzzpro (название последовательности начинается с MREB_). Если в списке находок окажется белок с другой мнемоникой, который не является MreB, это будет ложноположительная находка (FP). Для того чтобы рассчитать FN, необходимо знать, сколько всего белков MreB содержалось в файле /P/y24/term4/bacteria-sw.fasta. Ложноотрицательными будут те белки MreB, которые присутствуют в исходной базе белков, но не попали в итоговый отчет fuzzpro.
TP: 9
FP: 0
FN: 0
Точность поиска 100%
2. Поиск мотивов в белках программой MEME и поиск этих мотивов в банке
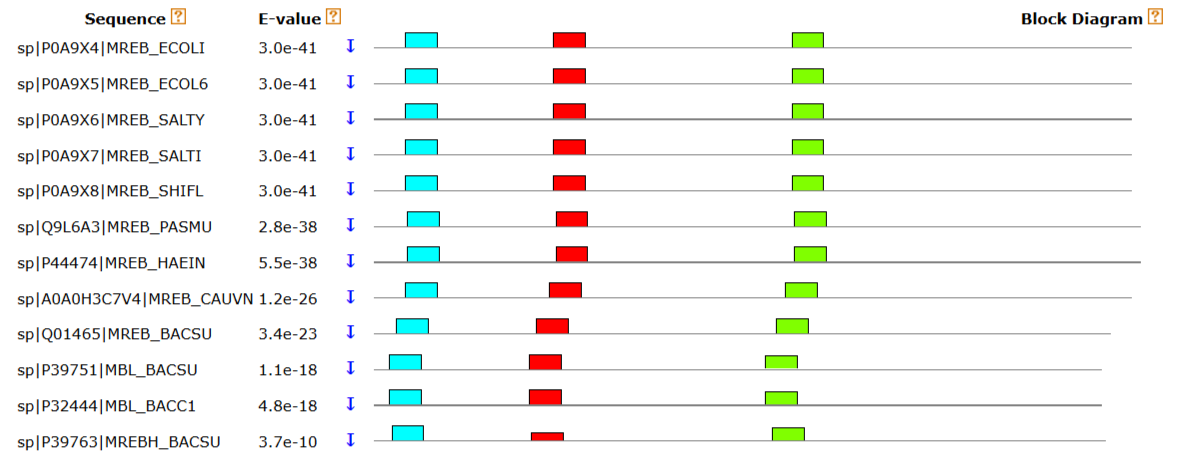
Теперь программой MEME найдём в тех же белках мотивы (опции: последовательности аминокислотные, по одному представителю мотива на последовательность, минимальная длина 8, максимальная длина 15, до трёх мотивов).
Мотивы нашлись во всех белках. Ни один из мотивов не соответствует ранее найденному мотиву. Для каждого из трех найденных мотивов MEME построил Logo (графическое представление консервативности). Если в позиции логотипа видна одна большая буква - она идет в паттерн как есть. Если букв несколько — они записываются в квадратные скобки [ ]. E-value последовательности отражает ожидаемое количество мотивов с таким же или более высоким информационным наполнением, которое можно найти в наборе случайных последовательностей того же размера. Значения для каждого из трёх мотивов низкие, следовательно, вероятность случайного возникновения таких паттернов практически невозможна. p-value - статистический показатель достоверности найденных мотивов для каждой конкретной последовательности. Оно показывает вероятность того, что случайная последовательность такой же длины будет иметь совпадения с найденными мотивами настолько же хорошие (или лучше), как и данная последовательность. Для всех последовательностей p-value низкие, это говорит о том, что такая комбинация мотивов практически не могла возникнуть случайно. Это подтверждает, что последовательности действительно относится к семейству белков, обладающих этими функциональными участками.


Программой MAST поищим в файле bacteria-sw.fasta найденные мотивы.
MAST нашёл 12 белков с похожим расположением мотивов, среди них добавились паралоги MreB - MBL и MreBH. После них идут последовательности с высокими значениями E-value. Также MAST нашел белки, где есть только 1 или 2 мотива, или они расположены в другом порядке — это могут быть функциональные аналоги MreB, которые имеют сходный активный центр, но другую общую структуру. В конце списка белки с E-value порядка 0.2 или выше. Это означает, что в них найдены лишь отдельные похожие участки, которые могут быть случайными совпадениями. Интересно, что MAST нашел сходство с белками-шаперонами DnaK. Это объясняется тем, что MreB и DnaK входят в одну суперсемью актиноподобных АТФаз и имеют отдаленно похожие каталитические домены. Таким образом, мотивы, полученнные meme, успешно нашли все основные белки MreB в базе, следовательно, мотивы достаточно точны, чтобы отличить MreB от других АТФаз.
3. Поиск последовательности Шайна — Дальгарно в геноме Borrelia garinii
Программой fuzznuc поищим ПШД в геноме Borrelia garinii.
На прямой цепи:Всего находок: 224
Длина генома: 993811
Количество A и G:grep -v ">" genome.fna | tr -cd 'G' | wc -c
Кол-во A = 355312; кол-во G = 138728
Частота A = 355312 / 993811 = 0.36; частота G = 138728 / 993811 = 0.14
Вероятность встретить мотив AGGAGG = (част. А)^2 * (част. G)^4 = 4.854*10^(-5)
Ожидаемое число находок на одной цепи = 4.854*10^(-5) * 993811 = 48
Тогда на двух цепях = 96.5
Наблюдаемое число находок (224) превышает ожидаемое (96,5) более чем в 2,3 раза. Следовательно вероятность того, что такое количество последовательностей Шайна — Дальгарно возникло случайно, очень мала. Это связано с давлением естественного отбора. Последовательность Шайна — Дальгарно критически важна для посадки рибосомы на мРНК и инициации трансляции. Геном намеренно сохраняет эти паттерны в промоторных областях генов.
Теперь, когда доказали, что последовательность не случайна, необходимо подтвердить её функциональность. Для этого нужно сопоставить 224 находки с геномной таблицей. Если находка расположена на расстоянии 5–15 нуклеотидов перед старт-кодоном аннотированного гена (CDS), она считается функциональной.
Для подсчёта процента находок, располагающихся в правильной позиции относительно старт-кодона какого-либо CDS, был написан python скрипт.
Всего находок: 224
В правильной позиции: 38 (16.96%)
Процент ПШД в правильной позиции небольшой, это может быть связано с разными причинами. Хотя окно 5–15 п.н. является классическим, у некоторых генов расстояние может быть чуть больше до 20 или меньше. Если взять расстояние 3-20 до старта, тогда 17.41%. У Borrelia garinii помимо ATG часто используются альтернативные старт-кодоны GTG и TTG. Также низкий процент может быть связан с высокой долей случайных G-богатых участков в АТ-богатом геноме или с использованием бактерией более коротких или вариативных версий последовательности Шайна — Дальгарно для части генов.