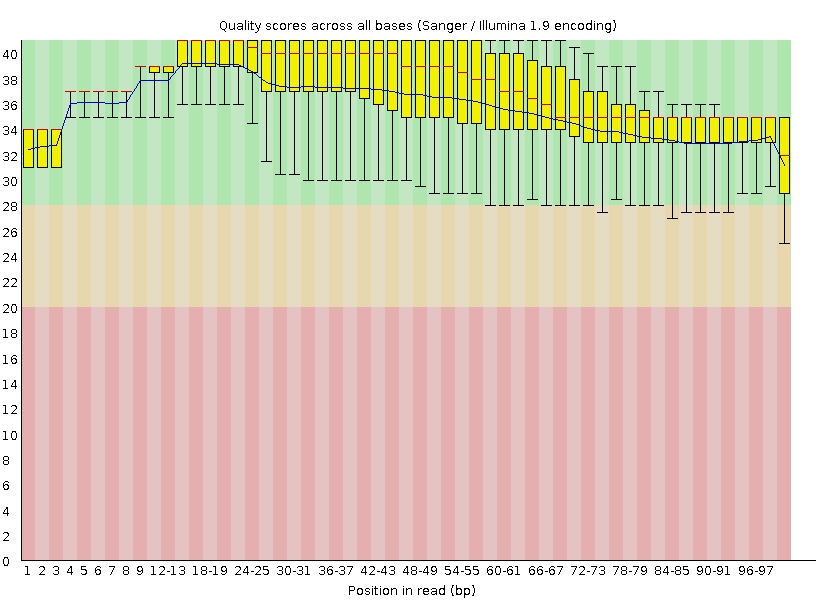
Необходимо было создать файл с полиморфизмами из нашего .bam файла. для этого была использовала команда
samtools mpileup -uf chr20.fasta -g -o poly.bcf out.bam
На выходе получился файл в формате .bcf. Далее, мы создавали файл со списком отличий между референсом и чтением с помощью команды:
bcftools call -cv -o differ.vcf poly.bcf
На выходе получился файл в формате .vcf. Опишем несколько полиморфизмов из данного файла:
1.Координаты:33961867
Тип полиморфизма:замена
Референс:T
Чтение:C
Глубина покрытия:19
Качество чтений:150.008
2.Координаты:33974207
Тип полиморфизма:замена
Референс:A
Чтение: G
Глубина покрытия:39
Качество чтений:207.009
3.Координаты:48524827
Тип полиморфизма:замена
Референс : A
Чтение: G
Глубина покрытия:41
Качество чтений:225.009
Далее мы производили аннотацию SNP с помощью программы annovar с использованием следюущих баз данных: refgene, dbsnp, 1000 genomes, GWAS, Clinvar.
Были использованы следующий команды(для поиска по указанным выше базам данных соответсвенно):
annotate_variation.pl -out refgen -build hg19 differ.annovar /nfs/srv/databases/annovar/humandb.old/
annotate_variation.pl -filter -out dbsnp -build hg19 -dbtype snp138 differ.annovar /nfs/srv/databases/annovar/humandb.old/
annotate_variation.pl -filter -dbtype 1000g2014oct_all -buildver hg19 -out 1000gen differ.annovar /nfs/srv/databases/annovar/humandb.old/
annotate_variation.pl -regionanno -build hg19 -out gwas -dbtype gwasCatalog differ.annovar /nfs/srv/databases/annovar/humandb.old/
annotate_variation.pl differ.annovar /nfs/srv/databases/annovar/humandb.old/ -filter -dbtype clinvar_20140211 -buildver hg19 -out clinvar
Число инделей: 0(в числе полиморфизмов только замены)
Число snp: 43
При аннотации по refseq мы получили 3 файла refgen.exonic_variant_function, refgen.log и refgen.variant_function. В файле refgen.variant_function мы видим, что
snp распределены по расположению(локализации): intronic(28), exonic(8), UTR5(1), intergenic(4), upstrem(2).
Гены, в которые попали наши snp:
UQCC1
GDF5
SPATA2
ZBP1
В файле refgen.exonic_variant_function содержится информация о синонимичности/несинонимичности замен нуклеотидов.
31 snp имеют rs
Аннотация по 1000 genomes показывают нам частоту аллелей.Наименьшая частота 0.0135783, наибольшая - 0.996406. В среднем частота = 0,303160174 (довольно высокая).
3 snp содержатся в GWAS, т.е. связаны с заболеваниями или предрасположенностью к заболеваниям.
Height(рост) chr20 34025756 34025756 A G hom 221.999 42
Psoriasis(псориаз) chr20 48522330 48522330 G A het 203.009 35
Hippocampal atrophy(атрофия гиппокампа) chr20 56190634 56190634 C T het 225.009 38
При аннотации по clinvar нашёлся один результат:
clinvar_20150629 CLINSIG=other;CLNDBN=Osteoarthritis_of_hip;CLNREVSTAT=no_assertion_criteria_provided;CLNACC=RCV000008898.2;
CLNDSDB=MedGen:OMIM:SNOMED_CT;CLNDSDBID=C0029410:612400:239872002 chr20 34025983 34025983 A G hom 182.007 10
Это означает, полиморфизм, имеющий координату 34025983, указан в базе данных OMIM(Менделевские наследования у человека) как отвечающий за остеоартрит тазобедренного сустава.
Сводная таблица по всем аннотациям