Занятия 11-12. Молекулярная динамика биологических молекул в GROMACS¶
Запуск молекулярной динамики, подготовка файлов.¶
В этом практикуме мы проводили моделирование самосборки липидного бислоя.
Исходные файлы:
%%capture
# на основе одного липида созадим ячейку с 64 липидами
!genconf -f dppc.gro -o b_64.gro -nbox 4 4 4
!editconf -f dppc.gro -o dppc1.pdb
!editconf -f b_64.gro -o dppc64.pdb
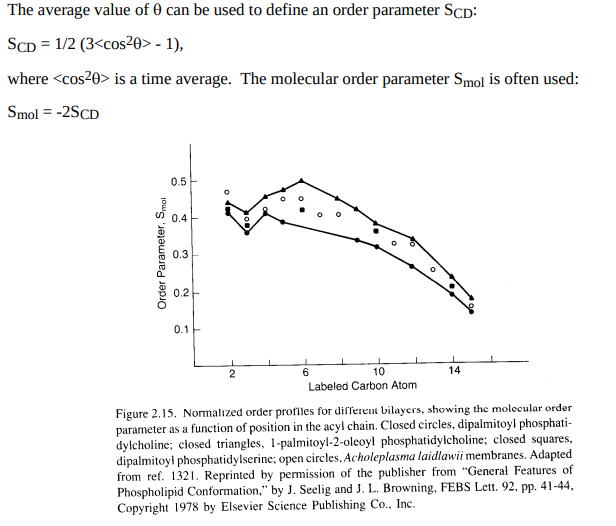
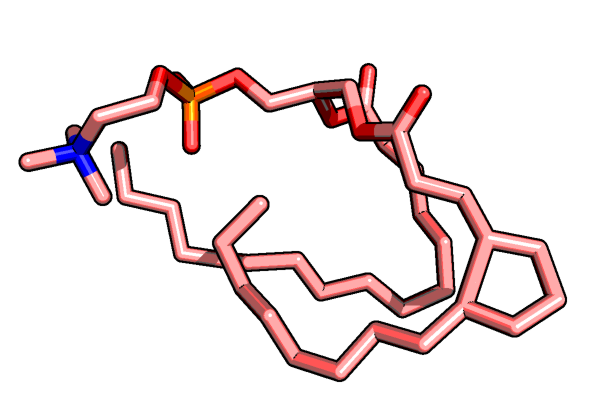 Рисунок 1. Структура липида DPPC.
Рисунок 1. Структура липида DPPC.
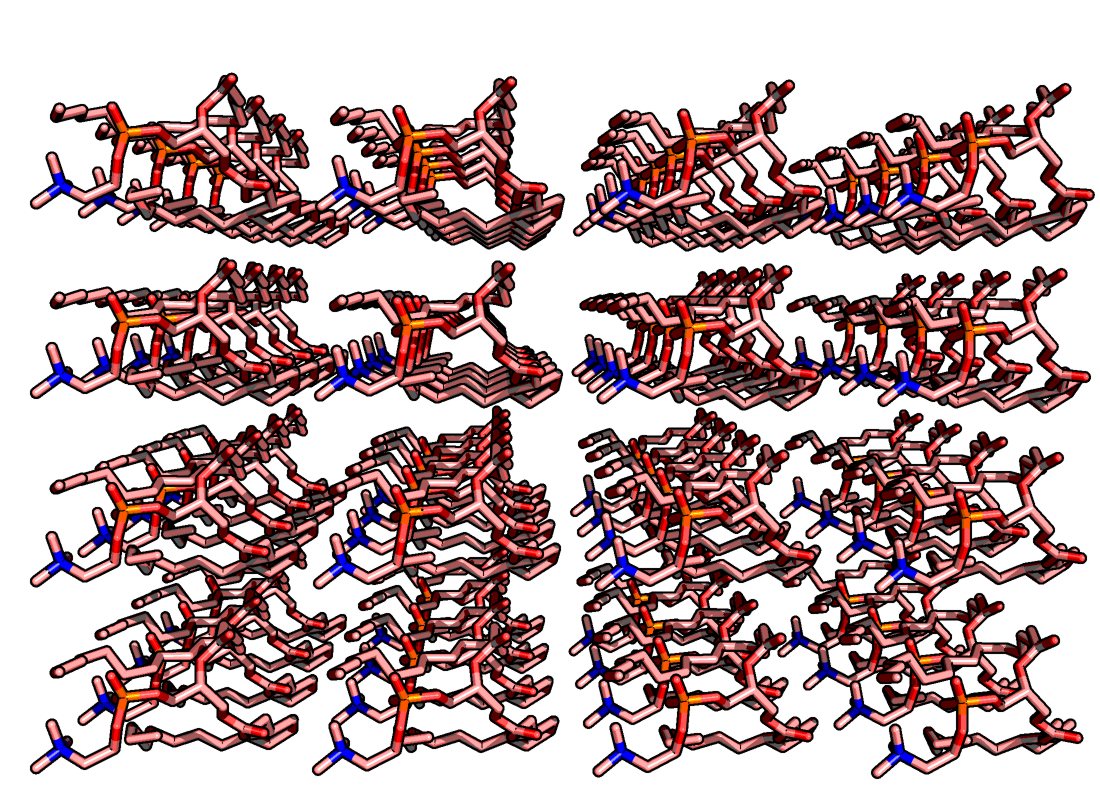 Рисунок 2. Ячейка из 64 липидов DPPC (4x4x4).
Рисунок 2. Ячейка из 64 липидов DPPC (4x4x4).
%%capture
# сделаем небольшой отступ в ячейке от липидов, что бы добавить примерно 2500 молекул воды
!editconf -f b_64.gro -o b_ec -d 0.5
# проведём оптимизацию геометрии системы, что бы удалить "плохие" контакты молекул
!grompp -f em -c b_ec -p b -o b_em -maxwarn 2
!mdrun -deffnm b_em -v
# Step= 0, Dmax= 2.0e-02 nm, Epot= 4.74007e+05 Fmax= 4.37970e+05, atom= 1842
# Step= 51, Dmax= 1.9e-06 nm, Epot= 3.20911e+03 Fmax= 6.16887e+02, atom= 2765
Стартовая максимальная сила 4.37970e+05, после оптимизации уменьшилась до 6.16887e+02.
%%capture
# добавим в ячейку молекулы воды типа spc
!genbox -cp b_em -p b -cs spc216 -o b_s
# проведём "утряску" воды
!grompp -f pr -c b_s -p b -o b_pr -maxwarn 1
!mdrun -deffnm b_pr -v
%%capture
!editconf -f b_s.gro -o b_s.pdb
!editconf -f b_pr.gro -o b_pr.pdb
 Рисунок 3a. Ячейка с 64 липидами с добавлением молекул воды.
Рисунок 3a. Ячейка с 64 липидами с добавлением молекул воды.
 Рисунок 3b. Ячейка с 64 липидами после утряски воды.
Рисунок 3b. Ячейка с 64 липидами после утряски воды.
Из рисунка 3 видно, что после утряски молекулы воды более плотно расположились внутри ячейки липидов и заняли пространства между липидами. До утряски вода лежит в ячейке упорядоченно, повторяющимися паттернами.
Далее на суперкомпьютере запускаем моделирование самосборки бислоя с помощью следующей команды:
sbatch -N1 --ntasks-per-node=2 -e error-gpu.log -o output.log
-t 350 -p gpu impi /opt/ccoe/gromacs-5.0.4/build/bin/gmx_mpi mdrun -testverlet -deffnm b_md -v
Анализ результатов.¶
Силовое поле, используемое при построении топологии - ffgmx
Заряд системы - 0 (в каждом липиде присутсвует аминогруппа (заряд +1) и фосфатная группа (заряд -1), которые компенсируют друг друга)
Размер и форма ячейки - 62.6x44.43x57.78 ангстрем, ортогональная, симметрия P1
Минимизация энергии (до добавления растворителя):
- Алгоритм минимизации энергии - steep
- Алгоритм расчёта электростатики и Ван-дер-Ваальсовых взаимодействий - Cut-off
- Модель, которой описывался растворитель - spc
Утряска растворителя:
- Число шагов - 1000
- Длина шага - 0.0002
- Алгоритм расчёта электростатики и Ван-дер-Ваальсовых взаимодействий - PME и Cut-off
- Алгоритмы термостата и баростата - V-rescale и no (без баростата)
Основной расчёт МД:
- Время моделирования - 50000 пс
- Число шагов - 10000000
- Длина шага - 0.005 пс
- Алгоритм интегратора - md
- Алгоритм расчёта электростатики и Ван-дер-Ваальсовых взаимодействий - PME и Cut-off
- Алгоритмы термостата и баростата - V-rescale и Berendsen
# сделаем PDB-файл с полученной динамикой
!echo 0 | trjconv -f b_md.xtc -s b_md.tpr -o b_pbc_1.pdb -skip 20 -pbc mol
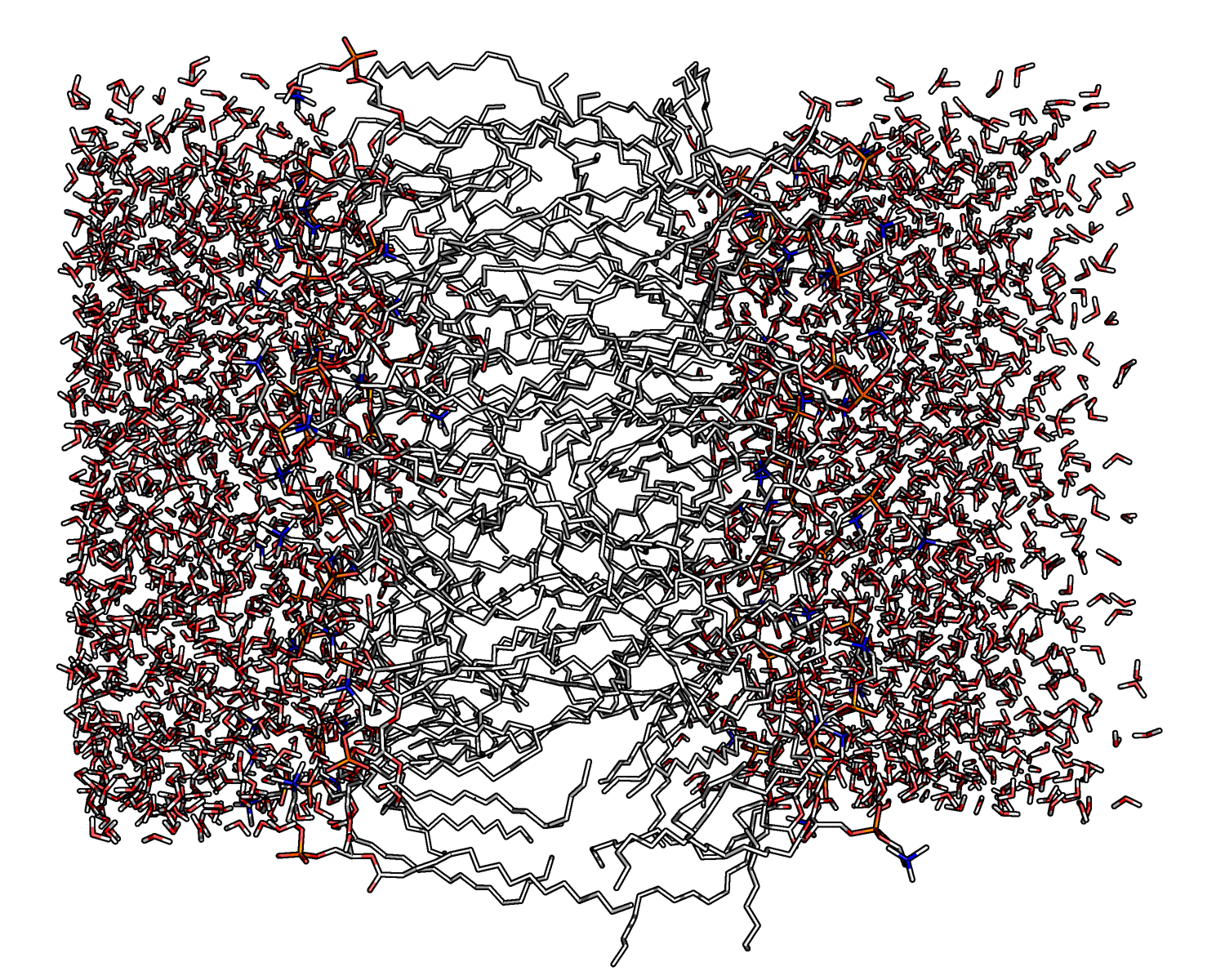 Рисунок 4. Липидный бислой после 9500 пс молекулярной динамики (модель №20).
Рисунок 4. Липидный бислой после 9500 пс молекулярной динамики (модель №20).
Уже на 10 модели (4500 пс) видны очертания бислоя, однако какие-то липиды все еще сидят внутри мембраны. Липиды полностю расходятся на 20 модели (9500 пс).
# определим размеры ячейки из траектории
!echo 0 | g_traj -f b_md.xtc -s b_md.tpr -ob box_1.xvg
В полученном файле - координаты ячейки в каждый момент времени.
Нормаль к бислою - ось X (скрипт).
Построим зависимость площади одного липида от времени.
coord = []
time = []
with open('box_1.xvg', 'r') as box:
lines = box.readlines()
for line in lines:
if line.startswith("@") or line.startswith("#"):
continue
else:
l = line.split('\t')
time.append(l[1])
coord.append(float(l[3])*float(l[4])/32)
%matplotlib inline
import numpy as np
import matplotlib.pyplot as plt
plt.figure(figsize=(10, 6))
plt.plot(time, coord, color="violet")
plt.xlabel('time')
plt.ylabel('lipid area, nm2')

Заметно, что площадь, занимаемая одним липидом, постепенно уменьшается. Резкое падение в начале объясняется формированием бислоя, далее бислой просто стабилизируется и уплотняется.
Здесь видно, что относительная стабилизация структуры наступает около 12000 пс. Ранее я указывала, что бислой собирается за 9500 пс, но, возможно, визуально сложно оценить, насколько полный и стабильный получается бислой. Поэтому из этого графика можно заключить, что бислой формируется чуть позже.
%%capture
!echo 3 3 | g_sas -f b_md.xtc -s b_md.tpr -o sas_b.xvg
time2 = []
hphob = []
hphil = []
with open('sas_b.xvg', 'r') as area:
lines2 = area.readlines()
for line in lines2:
if line.startswith("@") or line.startswith("#"):
continue
else:
m = line.split()
time2.append(m[0])
hphob.append(m[1])
hphil.append(m[2])
plt.figure(figsize=(10, 6))
plt.plot(time2, hphob, color="violet")
plt.plot(time2, hphil, color="orange")
plt.xlabel('time')
plt.ylabel('area SAS, ')
plt.legend(['hydrophobic', 'hydrophilic'])

В начале площадь гидрофобной поверхности очень большая, и в водной среде это энергетически невыгодно. При сборке бислоя площадь гидрофобной поверхности резко уменьшается и стабилизируется около 10000 пс. При этом сначала очень быстро формируется мицелла (на первых 10 моделях динамики), а затем уже она постепенно преобразуется в бислой. При образовании мицеллы резко увеличивается площадь гидрофильной поверхности (потому что вся поверхность стремится быть представленной гидрофильными головками липидов), затем она немного снижается при формировании бислоя.
 Рисунок 5. Подобие мицеллы, образующееся в начале моделирования (модель №3).
Рисунок 5. Подобие мицеллы, образующееся в начале моделирования (модель №3).
Теперь оценим меру порядка в начале и в конце тракетории.
%%capture
!g_order -s b_md -f b_md.xtc -o ord_end.xvg -n sn1.ndx -b 45000 -d X
!g_order -s b_md -f b_md.xtc -o ord_start.xvg -n sn1.ndx -e 5000 -d X
atom_start = []
order_start = []
with open('ord_start.xvg', 'r') as start:
lines3 = start.readlines()
for line in lines3:
if line.startswith("@") or line.startswith("#"):
continue
else:
n = line.split()
atom_start.append(n[0])
order_start.append(n[3])
atom_end = []
order_end = []
with open('ord_end.xvg', 'r') as end:
lines4 = end.readlines()
for line in lines4:
if line.startswith("@") or line.startswith("#"):
continue
else:
k = line.split()
atom_end.append(k[0])
order_end.append(k[3])
plt.figure(figsize=(10, 6))
plt.plot(atom_start, order_start, color="violet")
plt.plot(atom_end, order_end, color="orange")
plt.xlabel('atom #')
plt.ylabel('S (order parameter)')
plt.legend(['start', 'end'])

На этом графике изображена зависимость меры порядка (order parameter) от номера атома в гидрофобном хвосте. В первые 5000 пс липиды еще не упорядочены и перестраиваются для формирования мицеллы, а после и бислоя. Далее же заметно, что мера порядка убывает с номером атома. Это логично, так как хвосты липидов достаточно подвижны и остаются подвижными до самого конца динамики, а головки липидов плотно лежат в бислое.
Полученные результаты соответствуют наблюдениям, описанным в методичке. Также там указана формула для расчета S (меры порядка).