Занятие 5. Cеми-эмпирические и ab inito вычисления: Mopac и ORCA¶
Порфирин¶
import pybel
from os import system
import nglview as nv # для визуализации
import pandas as pd
# загружаем структуру порфирина по SMILES
mol = pybel.readstring('smi','C1=CC2=CC3=CC=C(N3)C=C4C=CC(=N4)C=C5C=CC(=N5)C=C1N2')
mol.addh() # добавляем водороды
mol.make3D(steps=1000) # оптимизируем (по умолчанию было 50 шагов)
mol.localopt(steps=500)
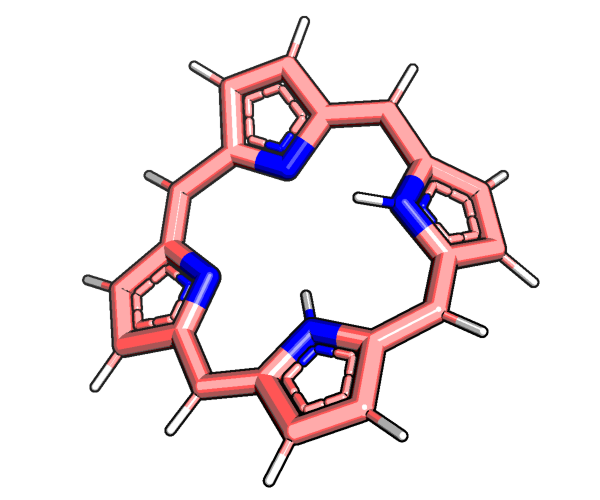 Рисунок 1. Структура порфирина до оптимизации.
Рисунок 1. Структура порфирина до оптимизации.
# функция для запуска MOPAC на kodomo, fn - название файла
def run_mopac(fn):
!export MOPAC_LICENSE='/home/preps/golovin/progs/mopac/'
!export LD_LIBRARY_PATH=${LD_LIBRARY_PATH}:/home/preps/golovin/progs/lib
!echo | /home/preps/golovin/progs/mopac/MOPAC2016.exe {fn}.mop
# просмотр результатов MOPAC как pdb-файла, fn - название файла
def read_mopac(fn):
opt = pybel.readfile('mopout', fn + '.out')
for i in opt:
i.write(format='pdb', filename = fn + '_mopac.pdb', overwrite=True)
return fn+'_mopac.pdb'
# оптимизация структуры порфирина PM6
mop_pm6 = mol.write(format='mopin', filename='porph_pm6.mop',
opt={'k':'PM6 CHARGE=%d' % mol.charge}, overwrite=True)
run_mopac('porph_pm6')
pm6 = read_mopac('porph_pm6')
#оптимизация структуры порфирина AM1
mop_am1 = mol.write(format='mopin', filename='porph_am1.mop',
opt={'k':'AM1 CHARGE=%d' % mol.charge}, overwrite=True)
run_mopac('porph_am1')
am1 = read_mopac('porph_am1')
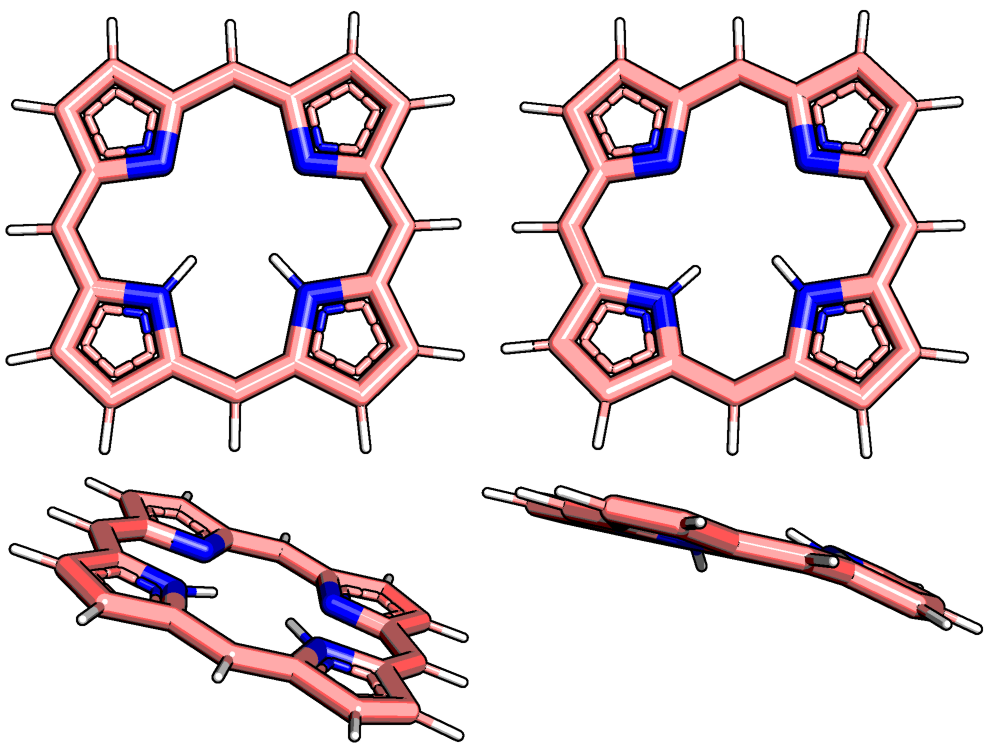 Рисунок 2. Структура порфирина после MOPAC: слева PM6, справа AM1.
Рисунок 2. Структура порфирина после MOPAC: слева PM6, справа AM1.
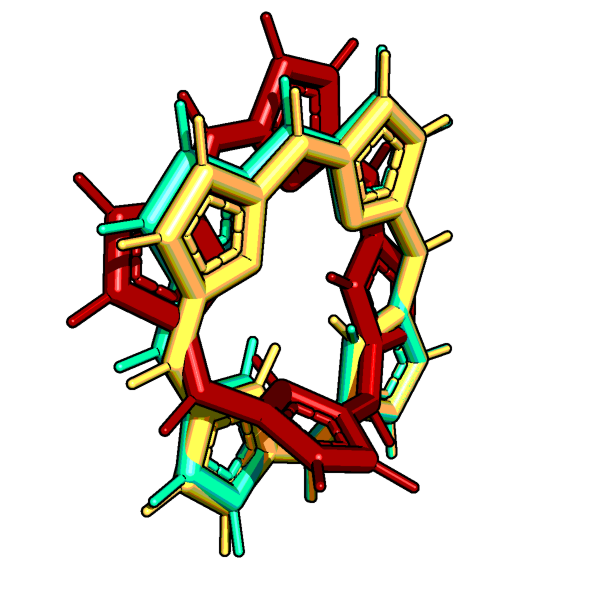 Рисунок 3. Наложение исходной структуры порфирина (красный) и структур, полученных в MOPAC способами PM6 (желтый) и AM1 (зеленый).
Рисунок 3. Наложение исходной структуры порфирина (красный) и структур, полученных в MOPAC способами PM6 (желтый) и AM1 (зеленый).
Из рисунков 1-3 заметно, что исходный порфирин был не совсем плоским. MOPAC исправляет эту проблему: способом PM6 получается абсолютно плоская структура, однако способ AM1 показывает не такой хороший результат: водороды в центре отклоняются от плоскости, и молекула слегка согнута пополам.
# создаем файл со структурой порфирина способом PM6 для генерации возбужденных состояний
with open('for_spectrum.mop', 'a') as excited:
excited.write("\ncis c.i.=4 meci oldgeo\n")
run_mopac('for_spectrum')
# нам нужны энергии полученных возбужденных состояний
! tail for_spectrum.out -n 25
У нас есть энергии для разных возбужденных состояний порфирина. По следующим формулам из энергий можно рассчитать длины волн, соответствующие переходам. $$E = h * \nu$$ $$\nu = \frac{c}{\lambda}$$ $$\lambda = \frac{hc}{E}$$ $$h=4,136 * 10^{−15} эВ*с$$ $$c = 3*10^8 м/с$$
en = [1.766222, 2.171501, 2.327997, 2.694919, 3.092498, 3.144815, 3.756781, 3.765370]
wl = []
for i in en:
l = 4.136 * 3 / i * 100
wl.append(l)
df = pd.DataFrame(data={'energy':en, 'wavelength':wl})
df
Тиминовый димер¶
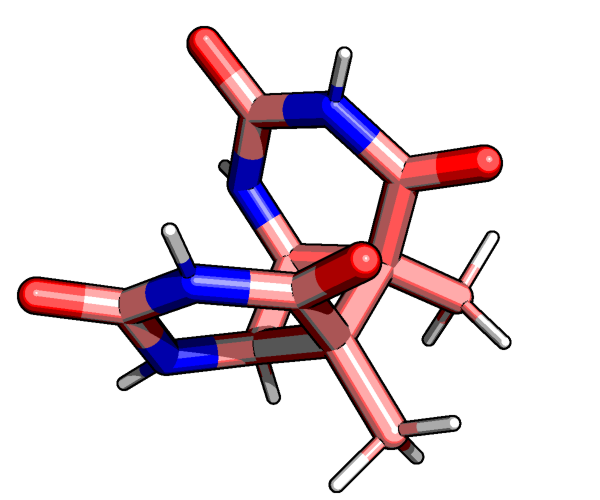 Рисунок 4. Структура тиминового димера.
Рисунок 4. Структура тиминового димера.
# функция для запуска MOPAC и просмотра результата как pdb-файла с заданным зарядом структуры
def mopac_td(fn, fn_new, charge):
fn.write(format='mopin', filename='%s.mop' % fn_new,
opt={'k':'PM6 CHARGE=%s XYZ' % charge}, overwrite=True)
run_mopac(fn_new)
opt = pybel.readfile('mopout','%s.out' % fn_new)
for i in opt:
i.write(format='pdb',filename='%s_mopac.pdb' % fn_new, overwrite=True)
%%capture
# оптимизация исходной структуры с зарядом 0 -> td_start
td = pybel.readfile('pdb', 'td.pdb').next()
mopac_td(td, 'td_start', '0');
%%capture
# оптимизация td_start с зарядом +2 -> td_charge
td_start = pybel.readfile('pdb', 'td_start_mopac.pdb').next()
mopac_td(td_start, 'td_charge', '+2')
%%capture
# оптимизация td_charge с зарядом 0 -> td_end
td_charge = pybel.readfile('pdb', 'td_charge_mopac.pdb').next()
mopac_td(td_charge, 'td_end', '0')
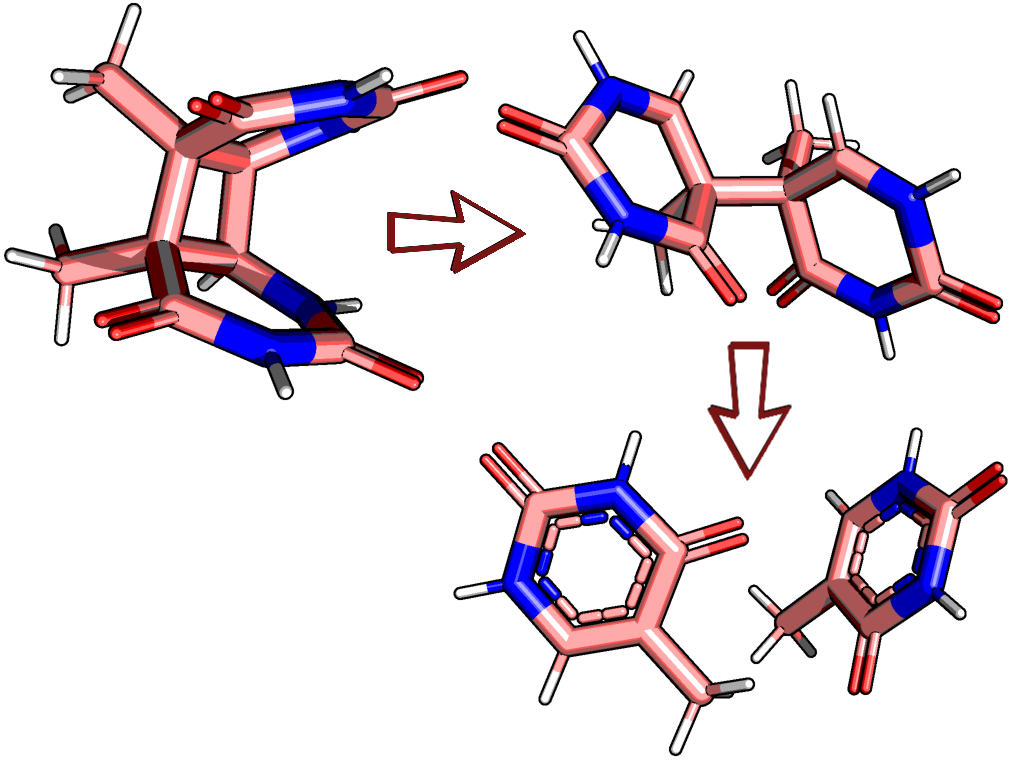 Рисунок 5. Изменение структуры тиминового тимера при оптимизации в MOPAC с зарядами 0, +2 и 0.
Рисунок 5. Изменение структуры тиминового тимера при оптимизации в MOPAC с зарядами 0, +2 и 0.
| Состояние | Энергия (total energy, эВ) |
|---|---|
| Исходное | -3273.57701 |
| Заряженное | -3253.90535 |
| Конечное | -3273.68570 |
Заряженное состояние соответствует локальному энергетическому максимуму (переходное состояние). Конечное состояние (отдельные тимины) немного ниже по энергии, чем исходное состояние (тиминовый димер), поэтому система сваливается в более выгодное состояние и не возвращается в состояние димера.
ORCA¶
def make_mol(smiles, name, ff):
mol = pybel.readstring('smi', smiles)
mol.addh()
mol.make3D(steps=2000, forcefield = ff)
#mol.localopt(steps=500)
mol.write(format="pdb", filename='%s.pdb' % name, overwrite=True)
mol.write(format='mopin', filename='%s.mop' % name,
opt={'k':'PM6 CHARGE=%d' % mol.charge}, overwrite=True)
run_mopac(name)
pdb = read_mopac(name)
struc = pybel.readfile('pdb', pdb).next()
# будем сравнивать методы HF (Хартри-Фока) и DFT (теорию функционала плотности)
for el in ('!HF RHF 6-31G','!DFT RHF 6-31G'):
method = el.split(' ')[0][1:]
for_orca = struc.write(format='orcainp',opt={'k':'%s' % el},
filename='%s_%s.oinp' % (name, method), overwrite=True)
with open('%s_%s.oinp' % (name, method),'r') as orca_file:
lines = orca_file.readlines()
with open('%s_%s.oinp' % (name, method),'w') as orca_input:
for line in lines:
if line.startswith("!"):
orca_input.write(el + '\n')
else:
orca_input.write(line)
%%capture
naph = make_mol('C1=CC=C2C=CC=CC2=C1', 'naph', 'mmff94')
azul = make_mol('C1=CC=C2C=CC=C2C=C1', 'azul', 'gaff')
В данном случае для азулена пришлось использовать другое силовое поле, потому что со стандартным полем (mmff94) он оптимизировался MOPAC в нафталин. При использовании поля gaff получается нормальная структура (рисунок 6).
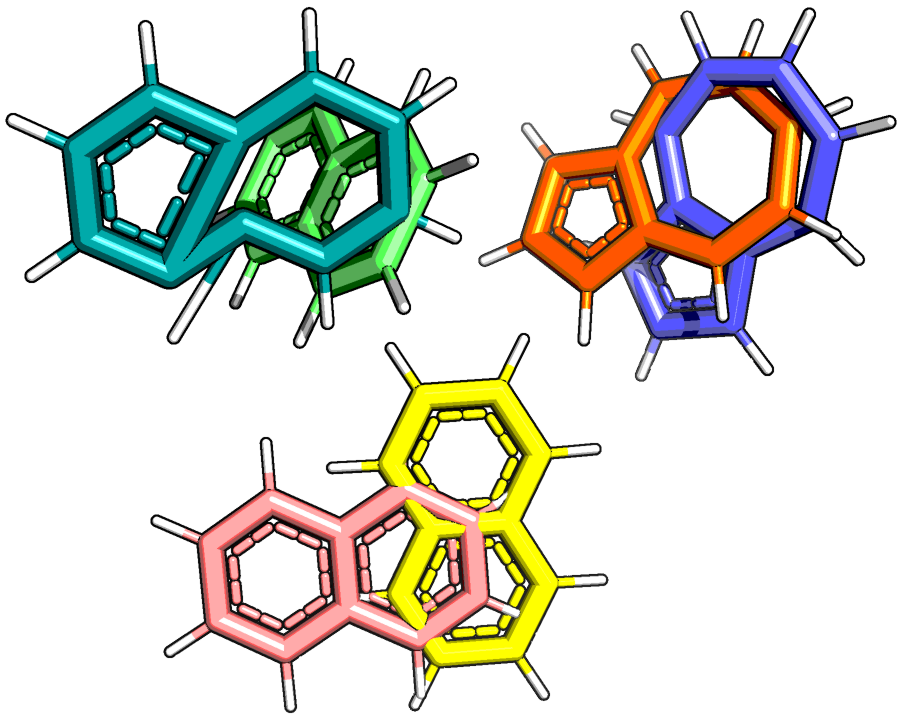 Рисунок 6. Структуры нафталина и азулена до и после оптимизации MOPAC. Сверху азулен: слева при использовании поля mmff94 (голубой - до оптимизации, зеленый - после), справа - при использовании поля gaff (оранжевый - до оптимизации, синий - после). Снизу нафталин (желтый - до оптимизации, розовый - после).
Рисунок 6. Структуры нафталина и азулена до и после оптимизации MOPAC. Сверху азулен: слева при использовании поля mmff94 (голубой - до оптимизации, зеленый - после), справа - при использовании поля gaff (оранжевый - до оптимизации, синий - после). Снизу нафталин (желтый - до оптимизации, розовый - после).
%%capture
!/srv/databases/orca/orca naph_DFT.oinp | tee orca-opt.log
# Total Energy -382.25561721 Eh -10401.70416 eV
%%capture
!/srv/databases/orca/orca naph_HF.oinp | tee orca-opt.log
# Total Energy -383.22014338 Eh -10427.95025 eV
%%capture
!/srv/databases/orca/orca azul_DFT.oinp | tee orca-opt.log
# Total Energy -382.19346390 Eh -10400.01288 eV
%%capture
!/srv/databases/orca/orca azul_HF.oinp | tee orca-opt.log
# Total Energy -383.14187968 Eh -10425.82058 eV
DFT = [-382.2556, -382.1935, 0.0621, 38.97]
HF = [-383.2201, -383.1419, 0.0782, 49.07]
# 1 Hartree = 627.509 kcal mol-1
df_na = pd.DataFrame(data={'DFT': DFT, 'HF': HF},
index=['E naph', 'E azul', 'ΔE, Hartree', 'ΔE, kcal/mol'])
df_na.T
Если известная энергия изомеризации нафталина в азулен составляет 35.3 ± 2.2 kCal/mol, то в данном случае метод DFT оказывается более точным.