Занятие 7. Изучение работы методов контроля температуры в молекулярной динамике.¶
%matplotlib inline
import numpy as np
import matplotlib.pyplot as plt
import time
tempc = ['be', 'vr', 'nh', 'an', 'sd'] # использованные алгоритмы температурного контроля
for i in tempc:
!grompp -f {i}.mdp -c etane.gro -p et.top -o et_{i}.tpr # создаем входные файлы для МД
!mdrun -deffnm et_{i} -v -nt 1 # МД
!echo 1 | trjconv -f et_{i}.trr -s et_{i}.tpr -o et_{i}.pdb # генерируем PDB
# оцениваем энергии (потенциальную и кинетическую)
!echo 10 11 0 | g_energy -f et_{i}.edr -o et_{i}_en.xvg -xvg none
# оцениваем длину C-C связи
!g_bond -f et_{i}.trr -s et_{i}.tpr -o bond_{i}.xvg -n b.ndx -xvg none
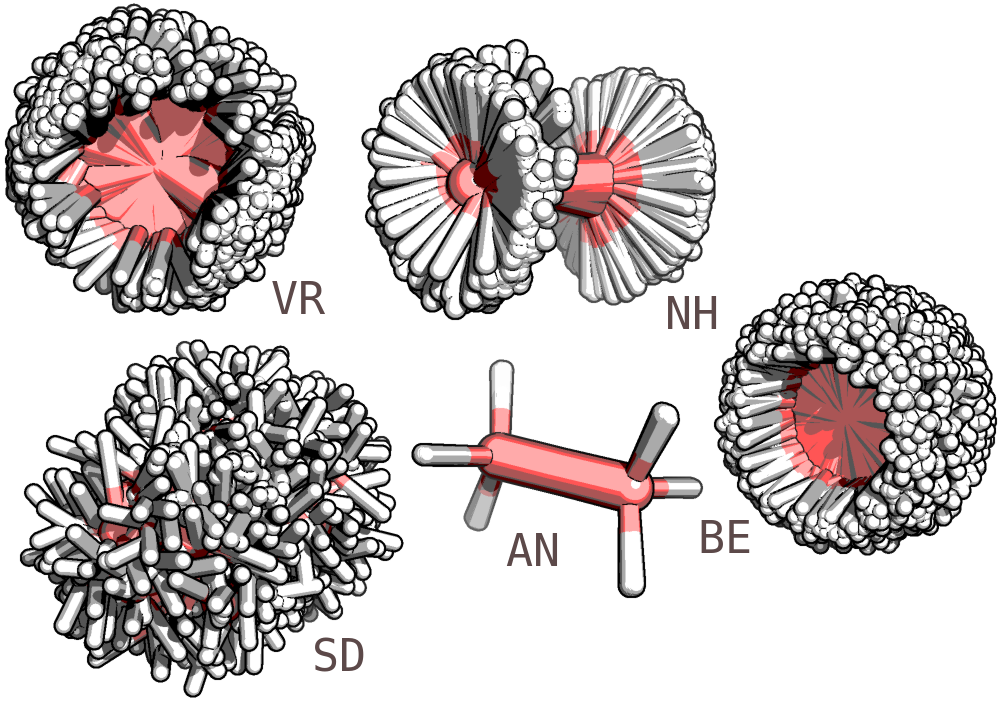 Рисунок 1. Полученные структуры этана (около 250 конформаций при МД) для пяти разных методов контроля температуры (подписаны на рисунке).
Рисунок 1. Полученные структуры этана (около 250 конформаций при МД) для пяти разных методов контроля температуры (подписаны на рисунке).
По рисунку 1 видно, что больше всего "дергается" этан в методе SD, а в методе AN практически ничего не изменяется (колеблется длина связи C-C и немного смещается положение атомов водорода). В методе VR также заметны достаточно сильные колебания.
# графики значений потенциальной и кинетической энергии для каждой конформации
# alg - обозначение алгоритма как в tempc (строка)
def energy_plot(alg):
tabs = np.loadtxt('et_' + alg + '_en.xvg')
x = tabs[:,0]
pot = tabs[:,1]
kin = tabs[:,2]
plt.figure(figsize=(10, 6))
plt.plot(x, pot, color='blue')
plt.plot(x, kin, color='violet', alpha=0.7)
plt.legend(['Potential', 'Kinetic energy'])
plt.title('Energies for the ' + alg + ' algorithm')
energy_plot('be') # метод Берендсена для контроля температуры

energy_plot('vr') # метод "Velocity rescale" для контроля температуры

energy_plot('nh') # метод Нуза-Хувера для контроля температуры

energy_plot('an') # метод Андерсена для контроля температуры

energy_plot('sd') # метод стохастической молекулярной динамики

Лучше всего выглядят методы Берендсена и Нуза-Хувера - к 250 конформации значения энергии стабилизируются и перестают жутко колебаться.
# распределения длин C-C связей в разных конформациях
# alg - обозначение алгоритма как в tempc (строка), w - ширина бина
def bondlen_plot(alg, w):
tabs = np.loadtxt('bond_' + alg + '.xvg')
x = tabs[:,0]
y = tabs[:,1]
plt.figure(figsize=(10, 6))
plt.bar(x, y, w, color='violet')
plt.title('C-C bond length distribution for the ' + alg + ' algorithm')
bondlen_plot('be', 0.0002)

bondlen_plot('vr', 0.0003)

bondlen_plot('nh', 0.0006)

bondlen_plot('an', 0.0002)

bondlen_plot('sd', 0.0005)

В целом, все полученные распределения имеют среднее около 0.153 нм, однако дисперсия наименьшая для методов Берендсена и Нуза-Хувера.
# оценка времени работы метода как среднее по 50 запускам для каждого из методов
def time_estimate(alg):
meantimes = []
for j in range(50):
s = time.time()
!grompp -f {alg}.mdp -c etane.gro -p et.top -o et_{alg}.tpr
e = time.time()
meantimes.append(e-s)
nptimes = np.array(meantimes)
npmean = np.mean(meantimes)
return npmean
times = {}
for i in tempc:
times[i] = time_estimate(i)
times
Все полученные времена работы практически одинаковы. Можно сказать, что быстрее всего работает метод Андерсена, но он дает самые неточные результаты. Не очень понятно, почему получились такие значения, потому что, по литературным данным, метод Нуза-Хувера более точный, но работает медленнее, чем метод Берендсена, а здесь такой разницы не наблюдается.
Для проверки того, какой из методов лучше, я также построила гистограммы для потенциальной энергии. В данном случае наиболее ярко выделяется метод Нуза-Хувера (см. график). Я бы сказала, что именно этот метод дает самые надежные результаты, хотя метод Берендсена также можно использовать (правда, на него ругается GROMACS и советует выбрать другой алгоритм).
# распределение потенциальной энергии
# alg - обозначение алгоритма как в tempc (строка), b - количество бинов
def energy_barplot(alg, b):
tabs = np.loadtxt('files/et_' + alg + '_en.xvg')
x = tabs[:,0]
en = tabs[:,1]
plt.figure(figsize=(10, 6))
plt.hist(en, b, color='blue')
plt.title('Potential energy distribution for the ' + alg + ' algorithm')
energy_barplot('nh', 50)
