Занятие 8. Гомологичное моделирование комплекса белка с лигандом¶
import sys
sys.path.append('/usr/lib/modeller9v7/modlib/')
sys.path.append('/usr/lib/modeller9v7/lib/x86_64-intel8/python2.5/')
import modeller
import _modeller
import modeller.automodel
import nglview as nv
%%capture
env=modeller.environ()
env.io.hetatm=True
%%capture
# COMPLEXES BETWEEN CHITOOLIGOSACCHARIDES AND LYSOZYME FROM THE RAINBOW TROUT
!wget http://www.pdb.org/pdb/files/1lmp.pdb
# Galleria mellonella (Greater wax moth) - P82174 (LYS_GALME)
!wget http://www.uniprot.org/uniprot/P82174.fasta
 Рисунок 1. Galleria mellonella (Greater wax moth) - большая восковая моль.
Рисунок 1. Galleria mellonella (Greater wax moth) - большая восковая моль.
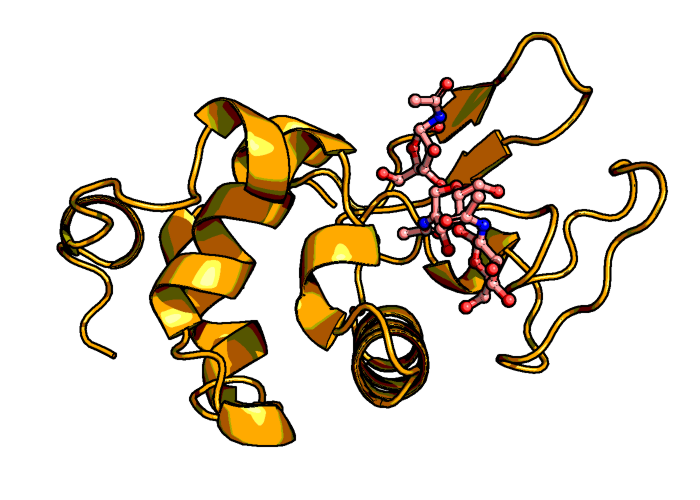 Рисунок 2. Исходная структура лизоцима радужной форели в комплексе с лигандом (PDB: 1LMP).
Рисунок 2. Исходная структура лизоцима радужной форели в комплексе с лигандом (PDB: 1LMP).
# функция для построения выравнивания и модели
# seq - название последовательности fasta, pdb - pdb-код белка-основы
# code1 - код для последовательности (query), code2 - код для pdb (target)
def align_mod(seq, pdb, code1, code2, align_name):
alignm = modeller.alignment(env)
alignm.append(file='%s.fasta' % seq, align_codes='all', alignment_format='FASTA')
## создадим модель
mdl = modeller.model(env, file='%s.pdb' % pdb, model_segment=('FIRST:'+'A', 'LAST:'+'A'))
## и добавим в выравнивание
alignm.append_model(mdl, atom_files='%s.pdb' % pdb, align_codes=pdb)
alignm[0].code = code1
alignm[1].code = code2
alignm.salign()
alignm.write(file='%s.ali' % align_name, alignment_format='PIR')
seq = alignm[0]
pdb = alignm[1]
a = modeller.automodel.automodel(env, alnfile='%s.ali' % align_name,
knowns = pdb.code, sequence = seq.code)
a.name = 'mod' + seq.code
a.starting_model = 1
a.ending_model = 5
a.make()
%%capture
align_mod('P82174', '1lmp', 'moth', 'trout', 'al')
# moth.B99990001.pdb 790.59070
# moth.B99990002.pdb 748.22253
# посмотрим на полученное выравнивание
!cat al.ali
И на полученные структуры.
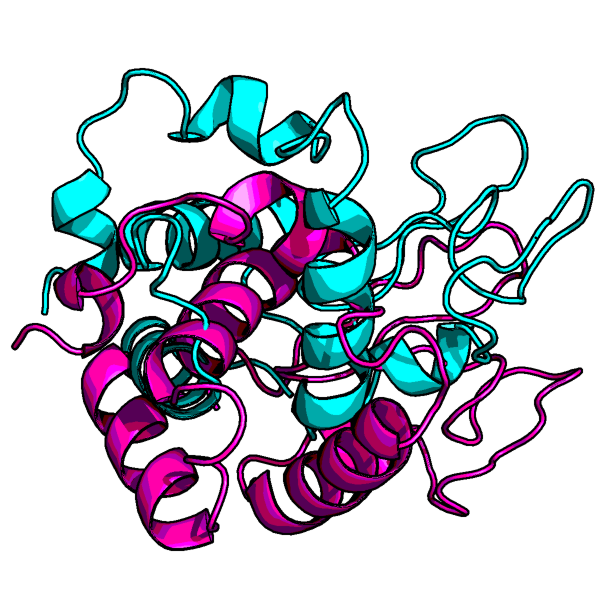 Рисунок 3. Синим показана первая модель, фиолетовым - последняя.
Рисунок 3. Синим показана первая модель, фиолетовым - последняя.
В последовательности и в структуре нашего белка из моли нет лиганда! Добавим лиганд:
new_seq = ''
for i in alignm[0].residues:
new_seq += i.code
new_seq += '...'
new_seq
with open('P82174_l.fasta', 'w') as new_fasta:
new_fasta.write('>P82174_ligand with NAG\n')
new_fasta.write(new_seq + '\n')
%%capture
align_mod('P82174_l', '1lmp', 'moth_lig', 'trout', 'align_lig1')
# moth_lig.B99990001.pdb 999.22644
# moth_lig.B99990002.pdb 1010.44019
 Рисунок 4a. Желтым показана первая модель, розовым - последняя. Лиганды покрашены соответственно.
Рисунок 4a. Желтым показана первая модель, розовым - последняя. Лиганды покрашены соответственно.
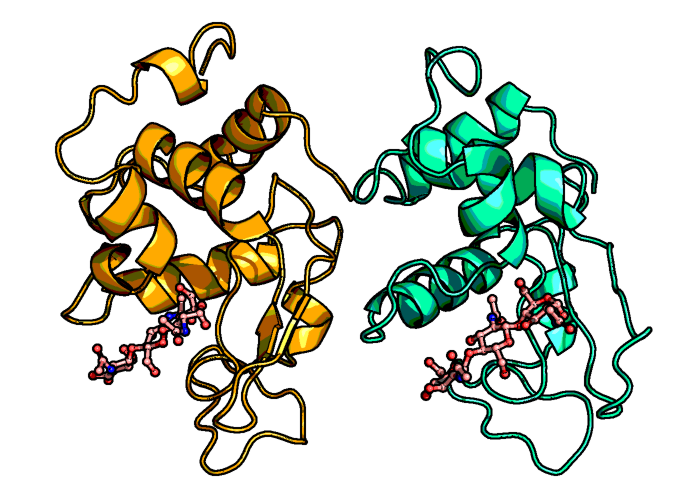 Рисунок 4b. Сравнение полученной модели (справа) со структурой исходного белка из форели (слева).
Рисунок 4b. Сравнение полученной модели (справа) со структурой исходного белка из форели (слева).
Из рисунка 4 видно, что полученная структура вполне соответсвует исходной: лиганд связывается в том же кармане, практически все $\alpha$-спирали совпадают (хотя их положение в пространстве и отличается), однако отсутствует $\beta$-лист.
# теперь попробуем изменить положение лиганда
from modeller import *
from modeller.automodel import *
class mymodel(modeller.automodel.automodel):
def special_restraints(self):
rsr = self.restraints
at = self.atoms
for x, y in [('CG:83','O6:228')]:
rsr.add(modeller.forms.gaussian(group = modeller.physical.xy_distance,
feature = modeller.features.distance(at[x],at[y]),
mean=3.0, stdev=0.1))
%%capture
align_mod('P82174_l', '1lmp', 'moth_lig2', 'trout', 'align_lig2')
# moth_lig2.B99990001.pdb 1059.81287
# moth_lig2.B99990002.pdb 978.77972
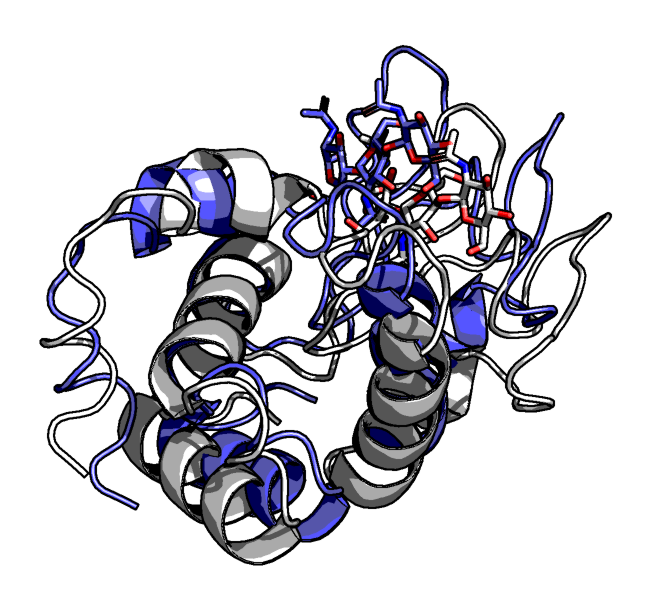 Рисунок 5a. Белым показана первая модель, синим - последняя. Лиганды покрашены соответственно.
Рисунок 5a. Белым показана первая модель, синим - последняя. Лиганды покрашены соответственно.
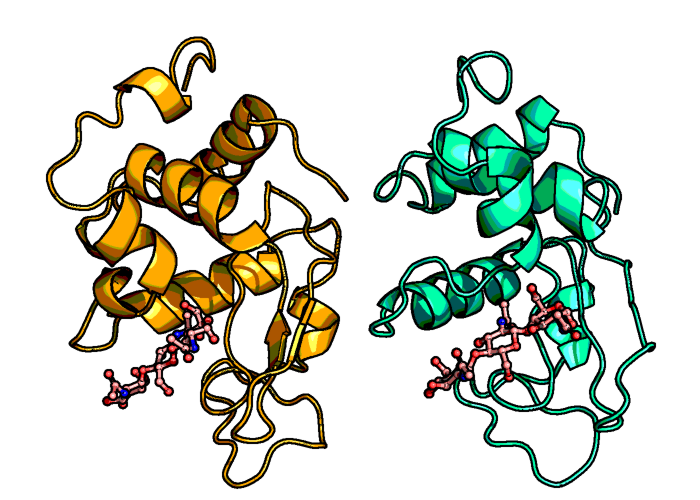 Рисунок 5b. Сравнение полученной модели (справа) со структурой исходного белка из форели (слева).
Рисунок 5b. Сравнение полученной модели (справа) со структурой исходного белка из форели (слева).
Теперь попробуем заменить все остатки в белке из моли на аланины.
ala_seq = ''
for i in alignm[0].residues:
ala_seq += 'A'
ala_seq += '...'
ala_seq
with open('P82174_ala.fasta', 'w') as ala_fasta:
ala_fasta.write('>P82174_ala with NAG\n')
ala_fasta.write(ala_seq + '\n')
%%capture
align_mod('P82174_ala', '1lmp', 'moth_ala_rsr', 'trout', 'align_ala_rsr')
# moth_ala_rsr.B99990001.pdb 909.90009
# moth_ala_rsr.B99990002.pdb 931.75018
В случае аланиновой последовательности у нас получается score 931.75, что несколько меньше, чем score для нормальной последовательности (1010.44). Несмотря на это, в структуре сильных отличий не наблюдается (рисунок 6).
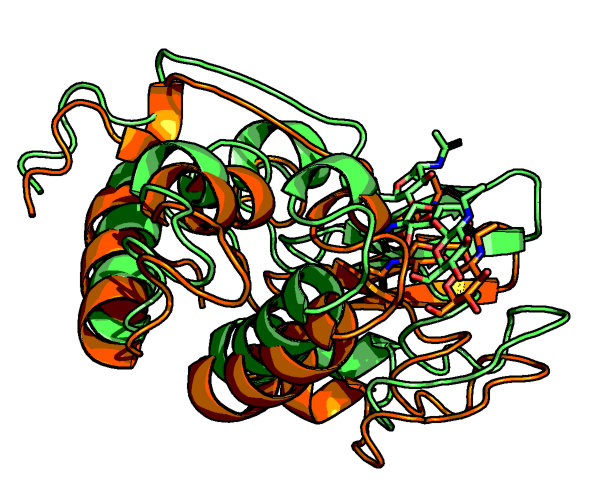 Рисунок 6a. Оранжевым показана первая модель, зеленым - последняя. Лиганды покрашены соответственно.
Рисунок 6a. Оранжевым показана первая модель, зеленым - последняя. Лиганды покрашены соответственно.
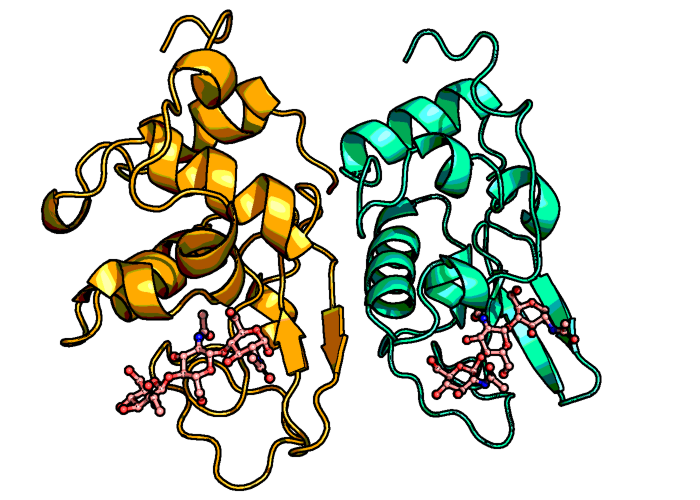 Рисунок 6b. Сравнение полученной модели (справа) со структурой исходного белка из форели (слева).
Рисунок 6b. Сравнение полученной модели (справа) со структурой исходного белка из форели (слева).
Заметно, что полученная структура немного отличается от предыдущих: $\alpha$-спирали имеют другую длину, появились $\beta$-листы (в том же месте, что и в белке из форели, но другой длины), лиганд также расположен немного по-другому. Однако структура в целом напоминает исходную.
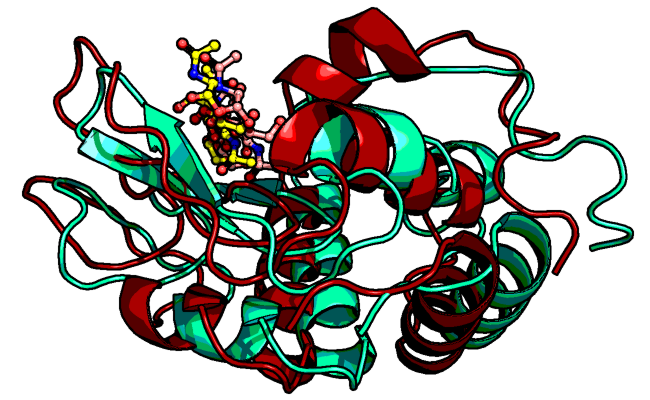 Рисунок 6c. Сравнение модели с нормальной последовательностью (красный с желтым лигандом) и с последовательностью из аланинов (зеленый с розовым лигадом).
Рисунок 6c. Сравнение модели с нормальной последовательностью (красный с желтым лигандом) и с последовательностью из аланинов (зеленый с розовым лигадом).
Наложение белков на рисунке 6c также помогает понять, как именно изменилась структура при замене всех аминокислот на аланины.