Сборка de novo
Для всех последующих действий использовались одноконцевые чтения РНК из хлоропластов Резуховидки Таля SRR1724089, полученные технологией Illumina
Задание 1. Подготовка чтений программой trimmomatic
Для начала нужно обрезать адаптеры с концов одноконцевых чтений, для этого нам нужны файлы с адаптерами в папке:
/mnt/scratch/NGS/adapters
В ней 6 файлов: NexteraPE-PE.fa, TruSeq2-PE.fa, TruSeq2-SE.fa, TruSeq3-PE-2.fa, TruSeq3-PE.fa, TruSeq3-SE.fa
У меня возник вопрос: стоит ли объединять адаптеры вместе, а если не объединять, то какие лучше использовать, ведь некоторые из них относятся к парным чтениям.
Если мы объединим все адаптеры в один файл и проведём триммирование с такими параметрами:
TrimmomaticSE -phred33 -threads 4 SRR1724089.fastq.gz SRR1724089_trimmed_all_adapters.fastq.gz ILLUMINACLIP:adapters.fasta:2:7:7
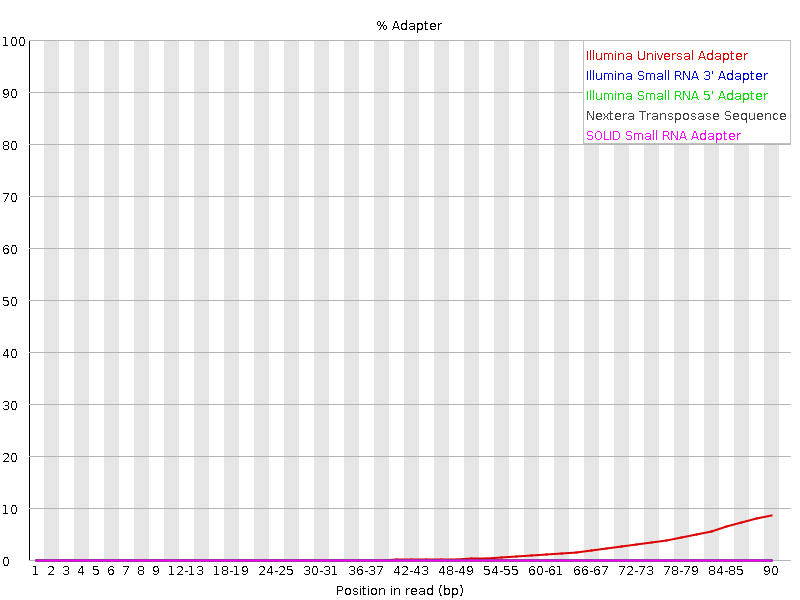
То в результате такой операции 8237 или 0.06% чтений отбросятся. Привожу файл с отчётом.
При этом fastqc анализ покажет, что количество адаптеров (Adapter Content) сократиться почти до нуля (или до нуля)
Однако мы также можем взять адаптеры только из файла TruSeq3-PE-2.fa и провести триммирование адаптеров командой:
TrimmomaticSE -phred33 -threads 4 SRR1724089.fastq.gz SRR1724089_trimmed_adapters.fastq.gz ILLUMINACLIP:TruSeq3-SE.fa:2:7:7
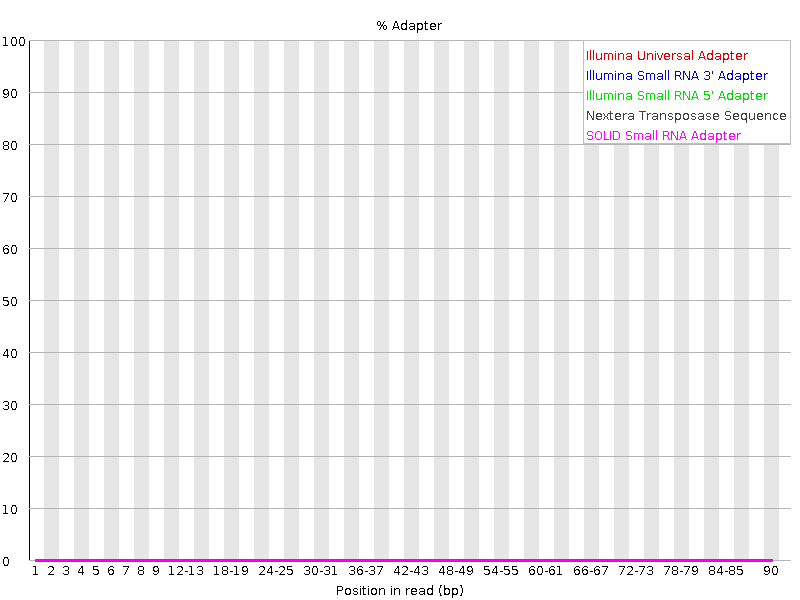
То у нас отбросятся только 1310 или 0.01% чтений (Отчет), при этом fastqc анализ также почти не выявит содержание адаптеров:
Возможно два вывода отсюда: либо когда мы используем все адаптеры отбрасываются лишние чтения из-за случайного совпадения, либо fastqc неправильно отображает количество адаптеров. Я доверюсь fastqc и буду использовать чтения, обрезанные при помощи адаптеров из TruSeq3-SE.fa. Не хочется терять данные. В пользу первой теории говорит тот факт, что файл с чтениями весит 943Мб, файл с вырезанными адаптерами из файла TruSeq3-SE.fa — 926Мб, а файл, из которого вырезали все адаптеры, — 925Мб
Затем к чтениям с обрезанными адаптерами я применил следующую команду:
TrimmomaticSE -phred33 -threads 4 SRR1724089_trimmed_adapters.fastq.gz SRR1724089_trimmed_adapters_quality.fastq.gz MINLEN:32 TRAILING:20
Она удаляет нуклеотиды с плохим качеством с 3' конца и фильтрует чтения длины меньше 32 нуклеотидов. В результате выполнения команды было отброшено 223135 или 1.54% чтений. Прилагаю файл с отчётом.
Начальный файл с чтениями весит 943МБ
Файл с отброшенными адаптерами весит 926Мб
Файл, дополнительно отфильтрованный по качеству и длине чтений, весит 899МБ
Я провёл анализ fastqc начального fastq файла и fastq файла после двух триммирований. И, действительно, заметно улучшение качества с 3' конца чтений:
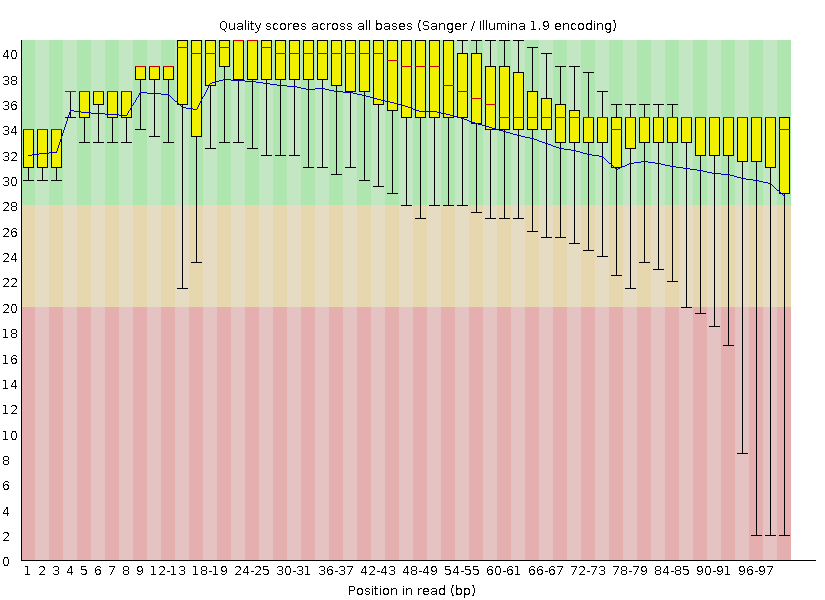
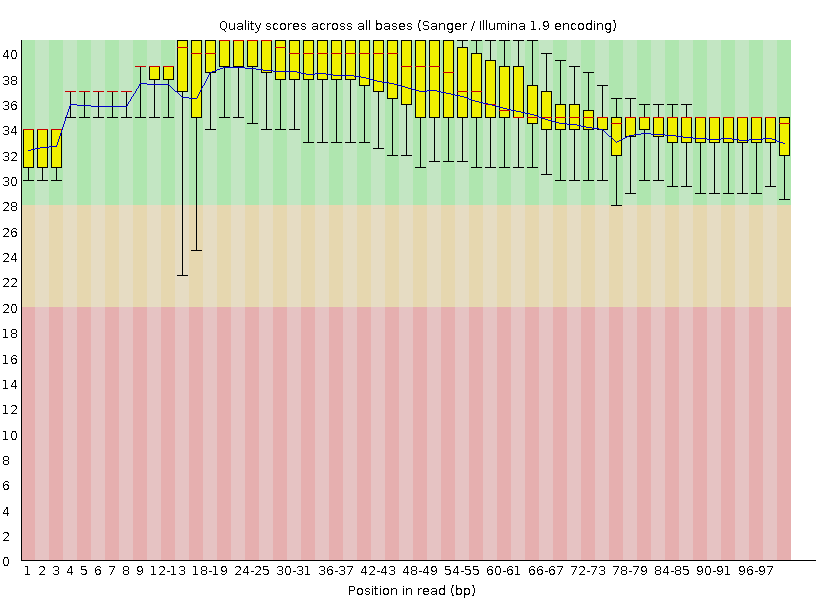
Задание 2. Подготовка k-меров на основе моих чтений
Для построения 31-меров для моих одноконцевых чтений длиной 101 нуклеотид я использовал следующую команду:
velveth velveth_out 31 -fastq.gz -short SRR1724089_trimmed_adapters_quality.fastq.gz
В результате выполнения команды внутри папки были созданы файлы Log (текстовый файл с параметрами запуска команд), Roadmaps (показывают расположение k-меров в чтениях) и Sequences (база данных всех чтений)
Задание 3. Сборка на основе генома
Затем, применяя к получившейся директории команду velvetg без параметров:
velvetg velveth_out
Я получил контиги генома хлоропласта. Используя, средство Python (Использованный код), я вычислил L50 и N50:
N50 = 124 нуклеотида
L50 = 18193 контига
Такие плохие качества сборки объясняются скорее всего тем, что в чтениях имелись как РНК хлоропласта, так и РНК ядра, это также становится очевидным, если мы посмотрим на три самых длинных контига и на их покрытие:
Контиг длиной 2348 нуклеотидов с покрытием 15.8
Контиг длиной 1801 нуклеотидов с покрытием 7.328628
Контиг длиной 1625 нуклеотидов с покрытием 19.47
Хотя при этом в сборке имеется контиг длиной 401 нуклеотид и покрытием 724. Очевидно, что первые три контига состоят из чтений ядра, а последний контиг состоит из чтений хлоропласта. Хлоропластный геном короткий и содержит много копий, поэтому очевидно, что при высоком кол-во чтений (в нашем случае >10 000 000) покрытие для контигов, принадлежащих геному хлоропласта, должно быть огромным. И так как мы хотим сборку именно хлоропластного генома, то должны сделать фильтр по покрытию, чтобы отобрать только те контиги, которые имеют высокий показатель покрытия.
С этой целья я добавил опции к команде velvetg:
velvetg velveth_new_out -exp_cov auto -cov_cutoff 70
-exp_cov auto позволяет программе определять ожидаемое покрытие для сборки, что может помочь в разрешении сложных ситуаций, -cov_cutoff 70 удаляет контиги с покрытием меньше 70.
Качество сборки значительно улучшилось:
N50 = 518 нуклеотидов
L50 = 74 контига
При это три самых больших контига имеют огромное покрытие:
Контиг длиной 5415 нуклеотидов с покрытием 694.8
Контиг длиной 3009 нуклеотидов с покрытием 607.3
Контиг длиной 2578 нуклеотидов с покрытием 221.77
Конечно имеются контиги с достаточно низким покрытием в 71-75, их длина колеблется от 110 до 260 нуклеотидов. Также имеются контиги небольшой длины (62-150 нуклеотидов), но имеющие огромное покрытие (больше 10 000, а иногда превышает даже 100 000).
При это стоит учесть, что если мы не так сильно ограничим покрытие контигов, то при выполнении следующей команды:
velvetg velveth_new_out -exp_cov auto -cov_cutoff 25
Мы сможем увидеть даже контиг длиной 8186 нуклеотидов, покрытие тоже будет приличным: 300
Кроме вышеупомянутого контига, здесь также имеются контиг длиной 5415 и покрытием 694.8 из предыдущей сборки, контиг длиной 3858 и покрытием 307.9, контиг длиной 2934 и покрытием 236.6
Задание 4. Гомология с геномом Arabidopsis thaliana
Для Megablast я использовал хлоропластную хромосому Arabidopsis thaliana из базы данных RefSec Genomes (идентификатор сборки генома GCF_000001735.4) с идентификатором NC_000932.1
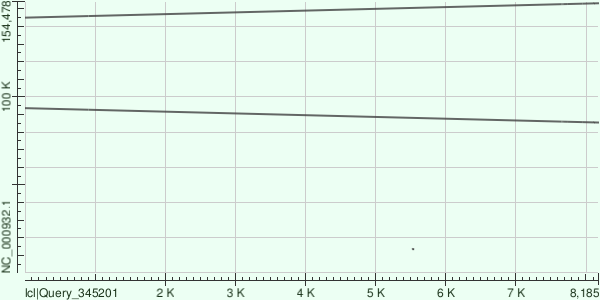
Я решил сначала пробластовать контиг длиной 8186 нуклеотидов на наличие гомологии с геномом Arabidopsis thaliana. Этот контиг выровнялся на три участка хлоропласта (файл с выравниванием), но интерес представляют только первые находки, так как они представляют идентичные выравнивания, но на разных цепях хромосомы хлоропласта. Первая находка на участке: от 93628 до 85453. Вторая находка на участке: от 145021 до 153196. Каждый участок отличается от запроса на 19 нуклеотидов и 9 гэпов и имеют один и тот же счёт: 15001 bits(8123). А расположение находок на противоположных цепях говорит нам об инвертированных повторах на участке от 85453 до 153196. Инвертированные повторы часто используются организмами для стабилизации генома и уменьшения скорости мутаций для жизненно важных генов
Score:15001, bits(8123), Expect:0.0, Identities:8166/8185(99%), Gaps:9/8185(0%) - характеристики выравнивания для каждой находки
В области выравнивания с контигов находятся: первый экзон гена рибосомального белка L2, ген рибосомального белка L23, тРНК изолейцина, ген белка Ycf2 (обеспечивает транспорт белков)
При этом контиг захватил только внешнюю часть инвертированного повтора. В подтверждение того, что мы имеем дело с инвертированными повторами привожу dot plot выравниваний:
Megablast не смог найти гомологии контига длиной 5415 нуклеотидов с геномом Arabidopsis thaliana, однако он нашёл почти полную гомологию с геномом бактериофага phiX174. В интернете было сказано, что геном этого бактериофага добавляется при секвенировании Illumina в библиотеку для внутреннего контроля (привожу файл с выравниваниями для доказательства). Так как этот контиг не принадлежит хромосоме хлоропласта, Dot plot приводить не буду.
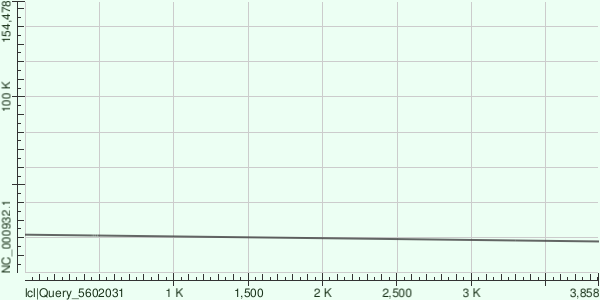
Затем я решил взять контиг длиной 3858 нуклеотидов из результатов выполнения команды velvetg при -cov_cutoff 25. Он также почти идеально выровнялся с участком на хромосоме хлоропласта с 21763 до 17910 нуклеотиды:
Score:6889 bits(7639), Expect:0.0, Identities:3847/3859(99%), Gaps:6/3859(0%), Strand: Plus/Minus
Данный участок соответствует генам бета субъединицы РНК-полимеразы. Привожу Dot Plot выравнивания:
Если мы возьмем контиг длиной 2934 нуклеотида (снова из результатов выполнения команды velvetg при -cov_cutoff 25) и пробластуем по хлоропласту, то получим прекрасное выравнивание по всей длине (файл) со следующими характеристиками:
Score:5601 bits(2913), Expect:0.0, Identities:2931/2935(99%), Gaps:2/2935(0%), Strand: Plus/Minus
по участку хромосомы хлоропласта от 25586 до 22653 нуклеотидов, соответствующему бета субъединице РНК полимеразы
Я также хотел бы взять контиг из результатов выполнения velvetg при параметре cov_cutoff 75 длиной 3009 нуклеотидов для поиска гомологии в геноме: он очень хорошо выровнялся (файл) по всей своей длине с хромосомой со следующими характеристиками:
Score:5544 bits(3002), Expect:0.0, Identities:3007/3009(99%), Gaps:1/3009(0%), Strand: Plus/Minus
по участку от 81103 до 78096 нуклеотидов, что соответствует генам: альфа субъединице РНК полимеразы, рибосомальным белкам: S11, L36, S8, L14
Также для контроля хорошо бы было взять контиг из результатов выполнения velvetg без всяких опций: я взял контиг длиной 2348 нуклеотидов. Как и следовало ожидать, megablast не смог найти гомологии с хромосомой хлоропласта, однако при поиске гомологии по всем хромосомам Megablast смог выровнять целых три участка из пятой хромосомы (файл) с моим контигом. При этом одна из находок выровнялась полностью с моим контигом без гэпов и мисматчей:
Показатели выравнивания самого крупного участка: Score:3278 bits(1775), Expect:0.0, Identities:1775/1775(100%), Gaps:0/1775(0%), Strand: Plus/Minus
участки выравниваний соответствуют экзонам гена GUN5, который кодирует субъединицу H магний-хелатазы, участвующую в синтезе хлорофилла