Практикум 10. HMM-профили и эволюционные домены
В качестве семейства я взял Ion transport protein PF00520. В этом семействе около 242 тысяч белков. Это семейство включает в себя каналы для ионов натрия, калия и кальция. Домен состоит из 6 спиралей. Селективность осуществляется за счёт P-петли между 5-ой и 6-ой спиралями.
В выравнивании 243 seed последовательностей и 258114 full последовательностей. Скорее всего количество последовательностей в выравнивании больше количества белков, так как один белок может содержать несколько доменов Ion transport protein.
В нём я выделил подсемейство: это белки птиц (класс Aves), у которых следующая доменная архитектура:
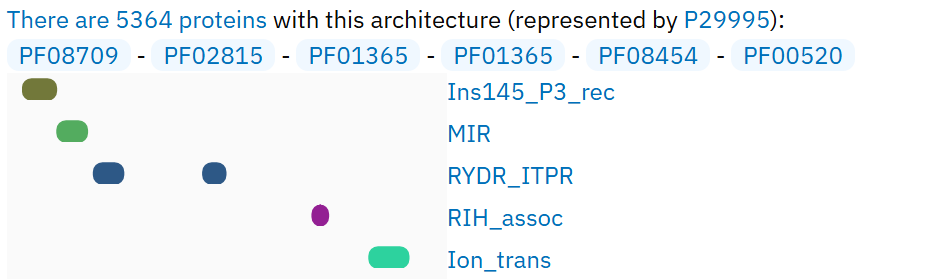
Белков птиц с доменом Ion transport protein - 33068,
а белков с данной данной доменной архитектурой среди всех таксонов 5364.
Мне нужно найти их пересечение, я не нашёл, как это сделать в PFAM. Поэтому я скачал последовательности белков обеих множеств и нашел их пересечение при помощи следующего кода Python. Получилось, что в подсемействе 867 белков.
Затем по доменам Ion transport protein в этих нужно было построить HMM-профиль. Однако сначала надо было выделить этот домен. Выравнивать огромное кол-во длинных белков заняло бы несколько часов, поэтому вручную это делать не хотелось. Поэтому я воспользовался hmmsearch. В качестве профиля взяль HMM-профиль эволюционного домена Ion transport protein.
Воспользовавшись следующей командой я получил находки доменов в белках:
hmmsearch --domtblout ion_hits.table PF00520.hmm aves_domain.fasta
Однако их кол-во оказалось больше кол-ва белков. Оказалось, что у некоторых белков кроме основной значимой находки имеется вторая находка (менее значимая - значение E-value больше 0.1). Поэтому я отсортировал все находки доменов по значимости и получил tsv-таблицу без комментариев:
awk '$12 < 1e-5' ion_hits.table > ion_hits_sorted.table
grep -v '#' ion_hits_sorted.table | sed -E 's/ +/\t/g' > ion_hits_sorted.tsv
Из этой таблицы я достал позиции доменов для каждого белка:
cut -f1,20,21 ion_hits_sorted.tsv | sed 's/|[^\t]*//g' > position_in_fasta.tsv
И при помощи следующего кода Python получил участки доменов белков.
Эти участки выровнял при помощи Muscle, а затем по данному выравниванию построил HMM-профиль:
hmmbuild --amino aves_5364.hmm aves_domain_seq_muscle.fasta
PFAM не позволил мне скачать все 242 тысячи Ion transport protein, поэтому я принял решение протестировать свой HMM-профиль не по всем белкам, а только по белкам птиц (которых, повторюсь, 33068 штук). И следующая команда это и осуществила:
hmmsearch -o final_hit.output --domtblout final_hit.table aves_5364.hmm protein-matching-aves.fasta
Если параметры поиска по умолчанию, то у нас получается следующая таблица:
| В подсемействе | Не в подсемействе | |
|---|---|---|
| Найдено | 867(все) | 1947 |
| Не найдено | 0 | 30254 |
Если же мы ограничим вес 325 (берем результаты у которых вес выше 325):
hmmsearch -T 325 -o final_fixed.output --domtblout final_fixed.table aves_5364.hmm protein-matching-aves.fasta
То получим следующие результаты:
| В подсемействе | Не в подсемействе | |
|---|---|---|
| Найдено | 867(все) | 130 |
| Не найдено | 0 | 32071 |
Я считаю данный вес оптимальным, так как он позволяет находить все нужные белки в подсемействе, но при этом сильно уменьшает кол-во ложных находок.
Если использовать немного другие формулировки, то получаем:
True positives: 867
False positives: 130
False negatives: 0
True negatives: 32071