Практикум 2. Филогенетическая реконструкция и сравнение дереевьев
Подготовка к построению деревьев:
Я создал список из идентификаторов цитохромов в Swiss-prot, затем по ним извлек их белковые последовательности в формате FASTA (файл) , затем выровнял при помощи алгоритма MUSCLE (файл), посмотрел выравнивание в jalview, все белки имели примерно одну и ту же длину и слишком длинных гэпов не было.
Затем при помощи скрипта из презентации конвертировал файл из формата fasta в phy (файл).
Построение деревьев
Первое дерево я построил при помощи программы fastme по алгоритму измерения расстояний p-distance, используя следующую команду:
fastme -i cyb.phy -pP -o cyb_p-distance.fastme
Второе дерево я построил снова при помощи программы fastme, но уже использовал алгоритм измерения расстояний — MtREV, поэтому применил вот такую команду:
fastme -i cyb.phy -pM -o cyb_MtREV.fastme
А третье дерево строилось программой iqtree с параметрами по умолчанию:
iqtree -s cyb.phy
Визуализация деревьев при помощи iTOL
На этом этапе я впал в ступор. Деревья визуализировались, однако нужно было выбрать, где его укоренить. Проблема в том, что два базальных вида Spiralia с мнемониками: BRAPC и PARGO из клады Gnathifera оказались отделены друг от друга. Я не знаю, насколько это хорошая идея, но я принял решения укоренить дерево за одного из них - PARGO. Для большего контроля я решил во всех деревьях сделать один и тот же корень, чтобы легче было сравнивать:
Приведу сначала скобочную формулу деревьев:
P-distance: (CYB_PARGO,((CYB_HETBL,(CYB_LUMTE,CYB_LOXAA)),(CYB_BUGNE,((CYB_MYZSE,CYB_LEPTH),(CYB_BRAPC,(CYB_MYTED,(CYB_CEPNE,CYB_ALBCA)))))))
MtREV: (CYB_PARGO,(CYB_HETBL,((CYB_LUMTE,CYB_LOXAA),(CYB_BUGNE,((CYB_BRAPC,(CYB_MYZSE,CYB_LEPTH)),(CYB_MYTED,(CYB_CEPNE,CYB_ALBCA)))))))
IQTree: (CYB_PARGO,((CYB_LOXAA,CYB_LUMTE),(CYB_MYTED,(CYB_HETBL,(CYB_BUGNE,((CYB_CEPNE,CYB_ALBCA),(CYB_BRAPC,(CYB_LEPTH,CYB_MYZSE))))))))
Теперь пройдемся по изображениям и сравним их с систематическим деревом, отметим ошибки реконструкции, а также правильные участки (их оказалось меньше, чем ошибок):
P-distance
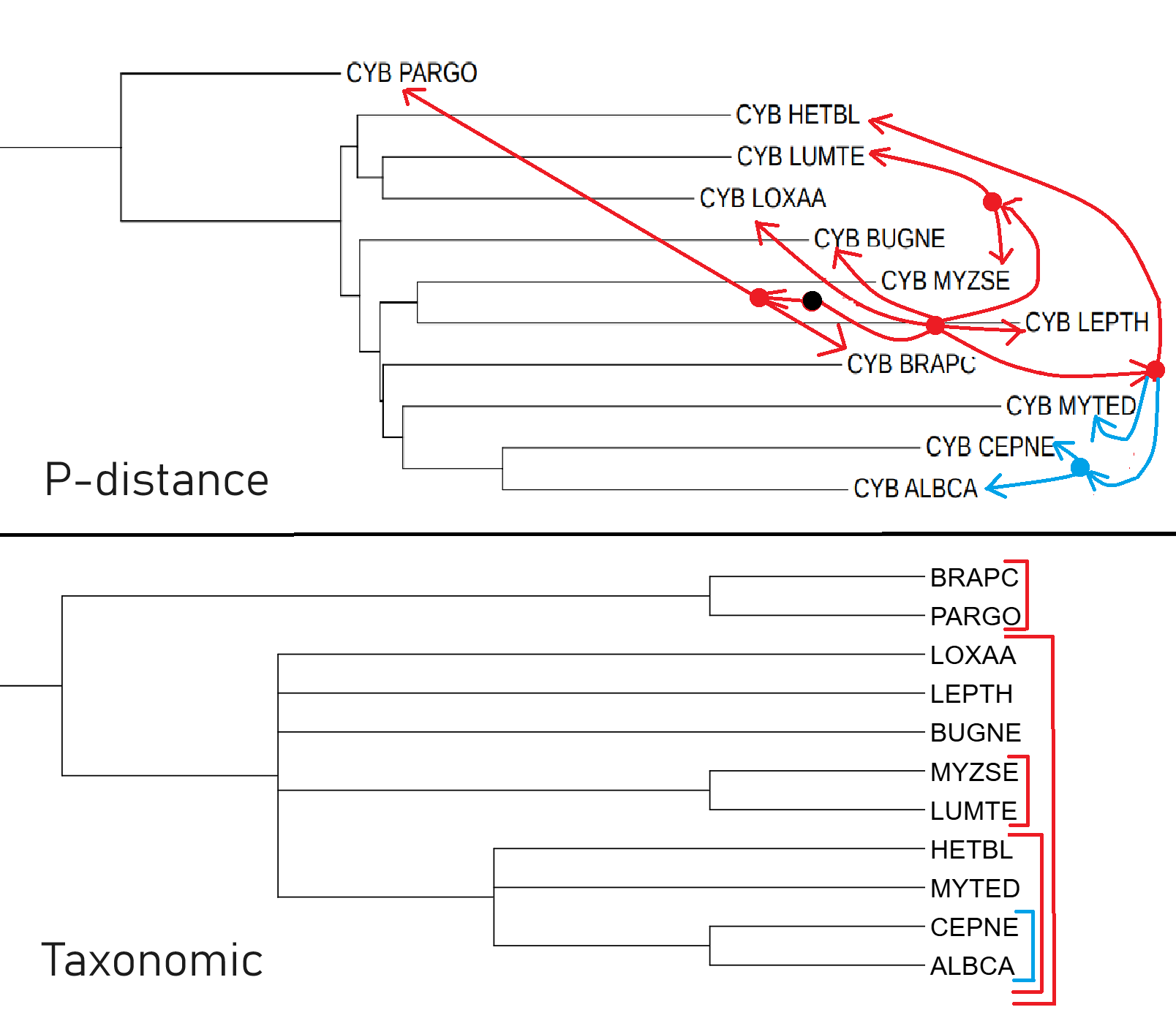
Поверх дерева P-distance я построил референсное дерево в виде стрелочек, чтобы было было легче их сравнивать (хотя выглядит запутанно). Синим цветом обозначил одинаковые ветви: они имеются и в таксономическом древе, и в референсном дереве
Красным цветом обозначил отличаюшиеся ветви: они есть в таксономическом дереве, но их нет в дереве p-distance
Стрелками обозначил направление от корня к листьям. Укоренение таксономического дерево я изобразил в виде черного круга, а точки ветвления синим или красным кругом (в зависимости от того, имеется ли это группа не дереве p-distance или нет)
Аналогичные действия я проделал для деревьев, построенных при помощи MtREV и IQTree.
Правильные участки P-distance: CEPNE и ALBCA в одной группе — род Helicina, MYTED с ( CEPNE и ALBCA ) в одной группе - тип Mollusca
Ошибки реконструкции: PARGO и BRAPC не находятся в одной базальной группе — Gnathifera, LUMTE находится в одной группе с LOXAA, а не с MYZSE (LUMTE и MYZSE принадлежат к типу Annelida), HETBL должен был находится рядом с MYTED, а не с LUMTE и LOXAA и т.д. Если кратко, то все организмы, кроме CEPNE, ALBCA и MYTED, сгруппированы неправильно.
MtREV
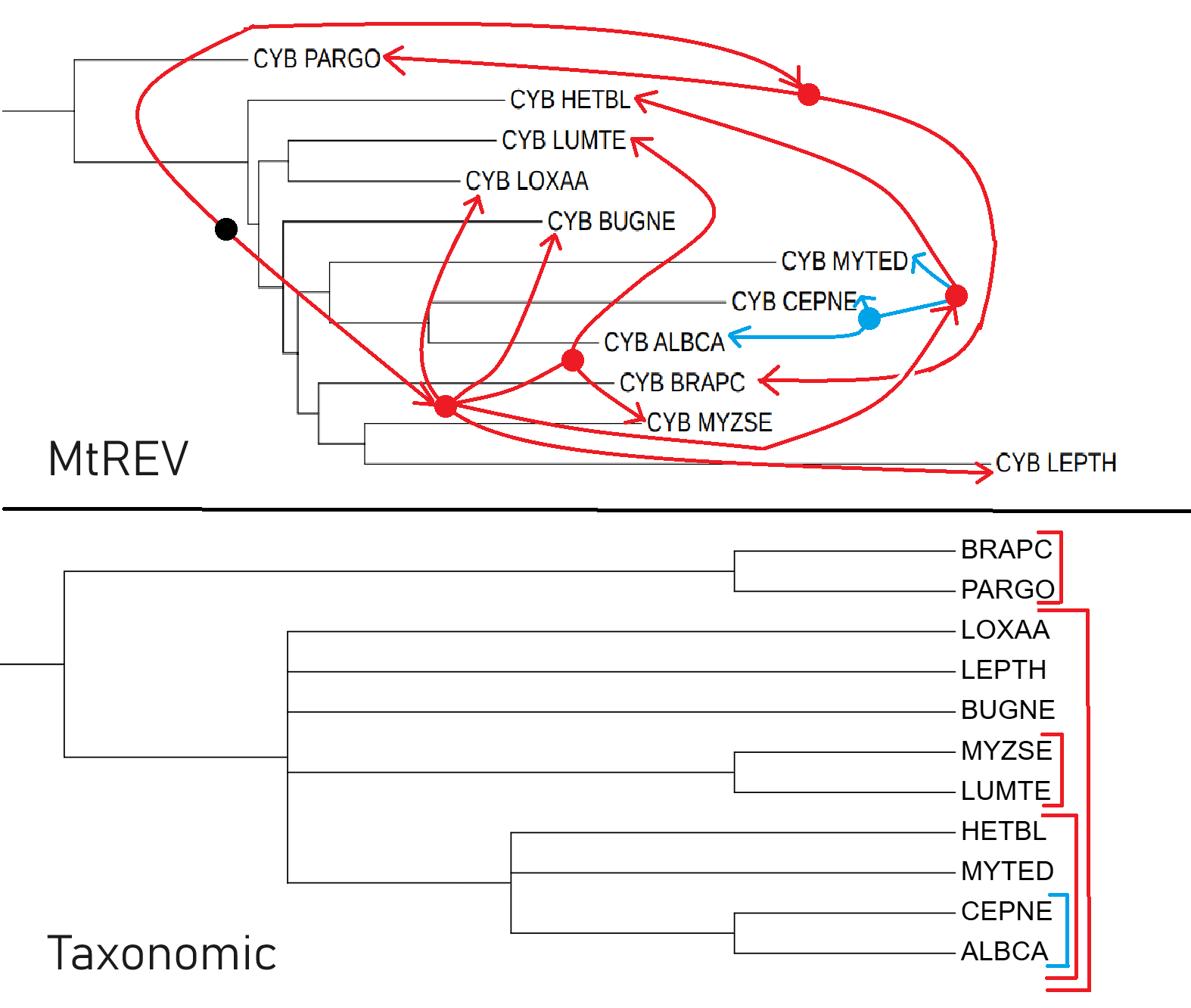
Дерево, построенное по MtREV, почти не отличается от дерева, построенного по P-distance. По-прежнему, единственный правильный участок: CEPNE, ALBCA и MYTED
От дерева построенного по P-distance отличается положением на дереве групп HETBL и BRAPC
IQTree
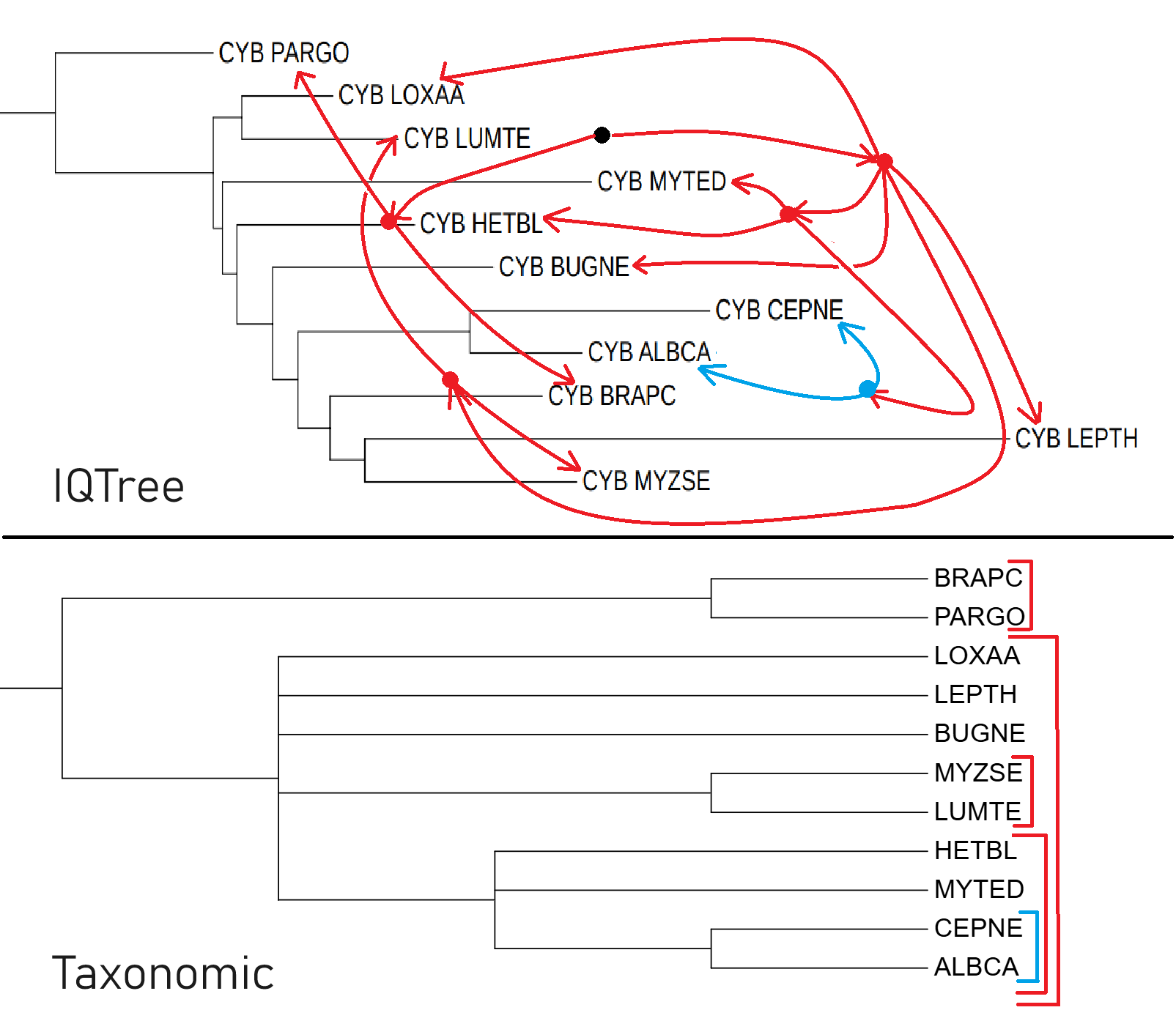
Дерево, построенное по IQTree отличается в более худшую сторону от предыдущих двух деревьев:
Единственная правильная группа: CEPNE и ALBCA.
MYTED же расположено неправильно и отходит от точки, близкой к корню дерева.
Неправильные группы: ( LUMTE с LOXAA ) и ( MYZSE с LEPTH ) сохраняются, HETBL имеет отличное от предыдущих двух леревьев положение, BRAPC расположен так же, как в дереве MtREV.
Выводы
В моём случае алгоритм построения дерева по консервативным аминокислотным последовательностям показал себя плохо
Возможные причины такого результата:
1) Исходное таксономическое дерево оказалось слишком неразрешённым, поэтому алгоритмы построения деревьев при такой выборке не справляются с задачей.
2) Построение деревьев по одному белку - изначально плохая идея. При построении деревьев должны учитываться и другие белки.
3) Виды в моей выборке оказались слишком далёкими, поэтому алгоритмы не смогли справиться и поделить их правильно на группы. Возможно имеет место быть конвергенция аминокислотных последовательностей, вследствие чего неродственные организмы стали иметь схожие последовательности цитохрома.