Построение деревьев по нуклеотидным последовательностям. Внешние группы. Бутстрэп
Построение деревьев по 12S рРНК
Я взял последовательности 12S (или small subunit) rRNA у организмов из предыдущих двух практикумов (FASTA)
Затем выравнял их алгоритмом Muscle. Но передо мной встал вопрос, как мне учитывать гэпы? Полностью убрать? Оставить всё на волю программе? Или убрать столбцы, где гэпов больше 70%?
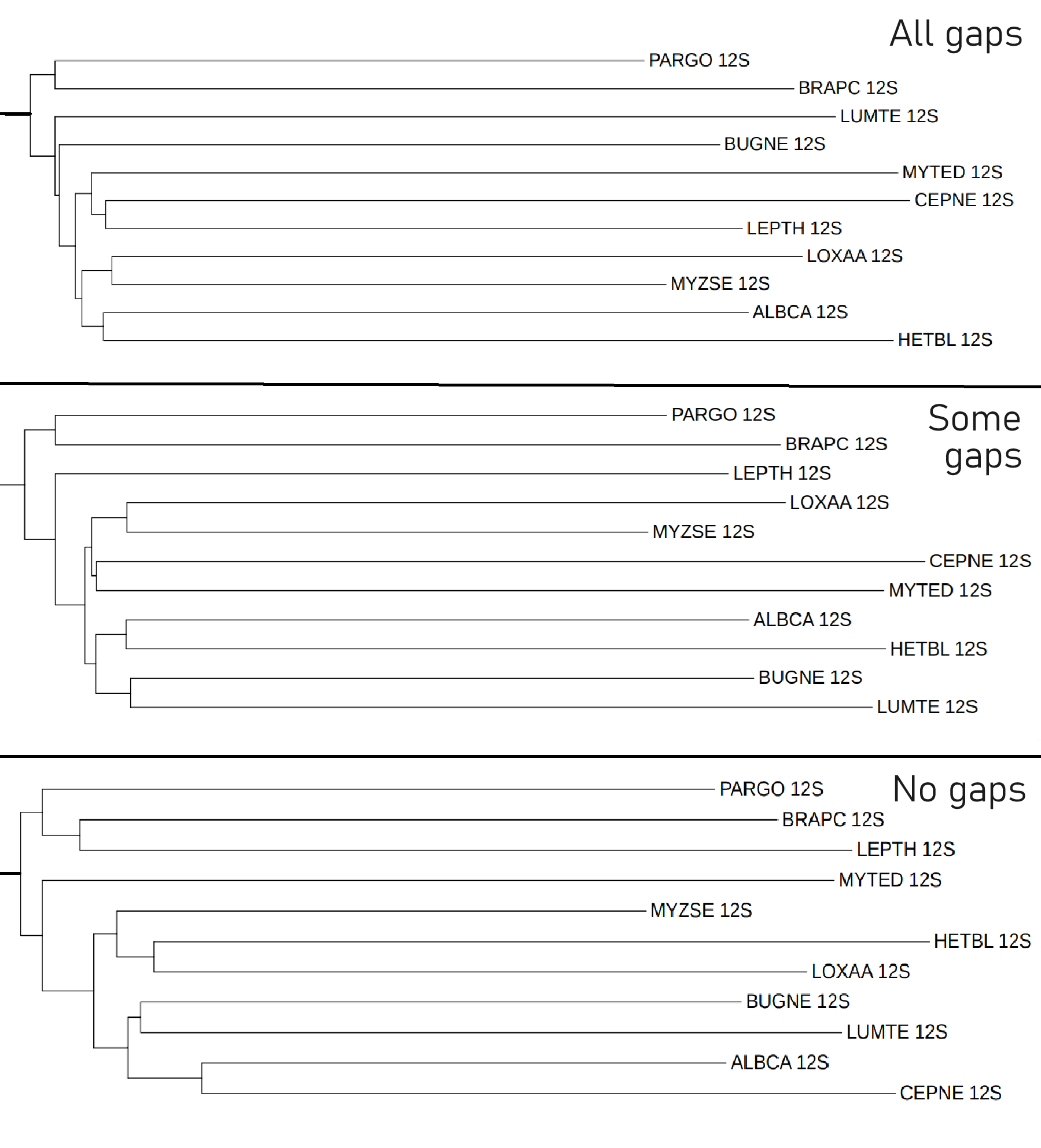
Чтобы это выяснить, давайте сравним деревья с разным количеством гэпов:
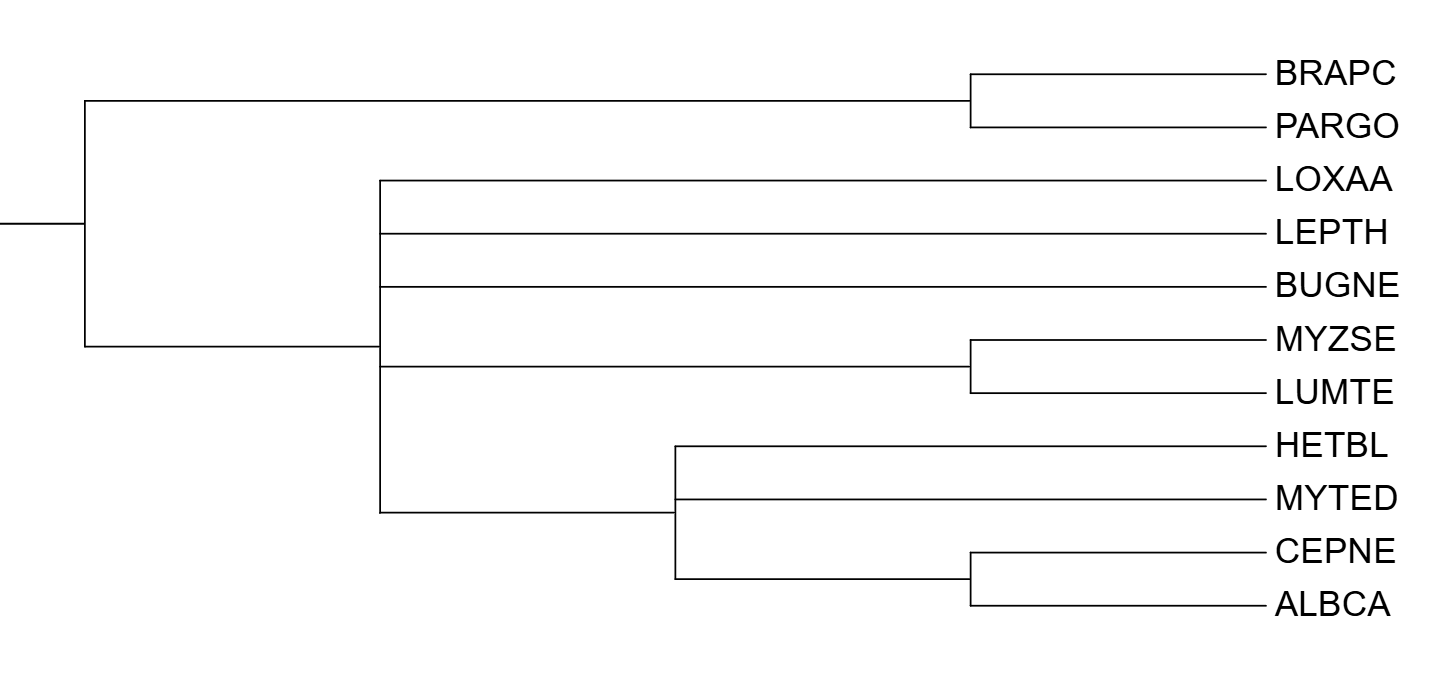
Снизу приведу референсное (таксономическое) дерево для сравнения с ним:
Деревья 'Some gaps' и 'All gaps' смогли правильно выделить правильно группу Gnathifera
Но дерево 'No gaps' почему-то добавило к этой группе организм с мнемоникой LEPTH, который относится к Lophotrochozoa, а не Gnathifera.
Однако дерево без гэпов всё же смогло объединить между собой группы CEPNE и ALBCA.
Также из положительных, но не абсолютно правильных можно выделить несколько черт:
– дерево 'All gaps' объединило родственные HETBL и ALBCA в одну группу
– дерево 'Some gaps' объединило родственные HETBL и ALBCA в одну группу, вынесло LEPTH за пределы всех остальных Lophotrochozoa
Самым худшим, по моему мнению, оказалось дерево 'No gaps', а самым (относительно) лучшим - 'Some gaps'. Поэтому именно его я буду использовать для сравнения с деревом построенным по цитохрому B
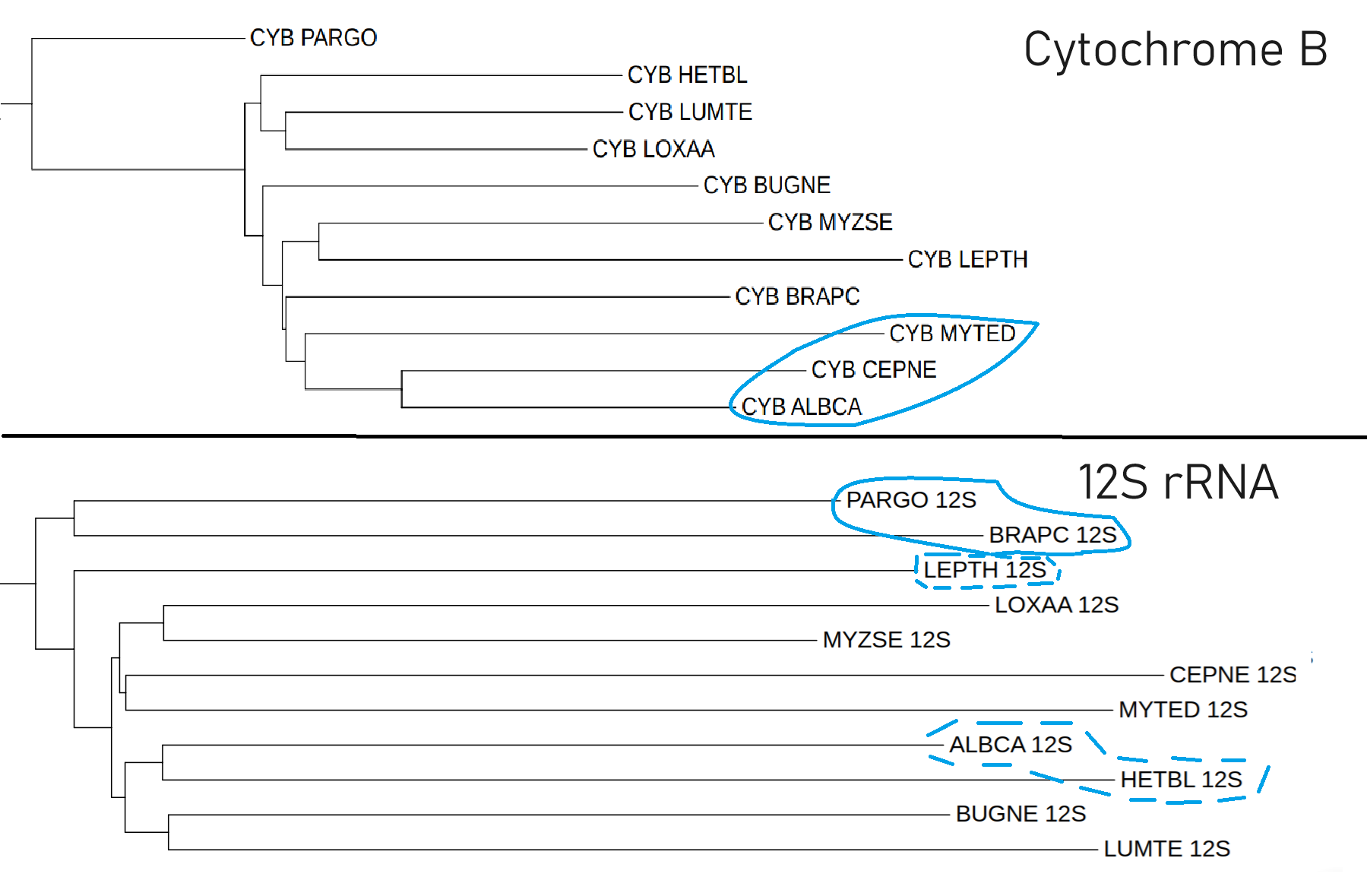
Исходя из третьего рисунка можно прийти к выводу, что для моих организмов деревья, построенные по нуклеотидным последовательностям 12S rRNA, смогли лучше определить родство для дальних родственников и отделить базальную группу (Gnathifera – BRAPC и PARGO) от всех остальных видов, однако не смогли определить группу близкородственных организмов (род Helicina – CEPNE + ALBCA). Для деревьев, построенных по белковым последовательностям цитохрома B, верно обратное.
Однако оба дерева не смогли объединить MYZSE и LUMTE в группу Annelida, показать отдаленность от всех остальных видов LOXAA, LEPTH и BUGNE, а также полностью реконструировать группу Mollusca, состоящую из HETBL, MYTED и CEPNE с ALBCA.
Также для интереса я построил по нуклеотидной последовательности 12S деревья при помощи эволюционных моделей Kimura и Symmetric, вы можете посмотреть их здесь (папка с картинками)
Крайне неплохим получилось дерево, построенное программой IQtree:
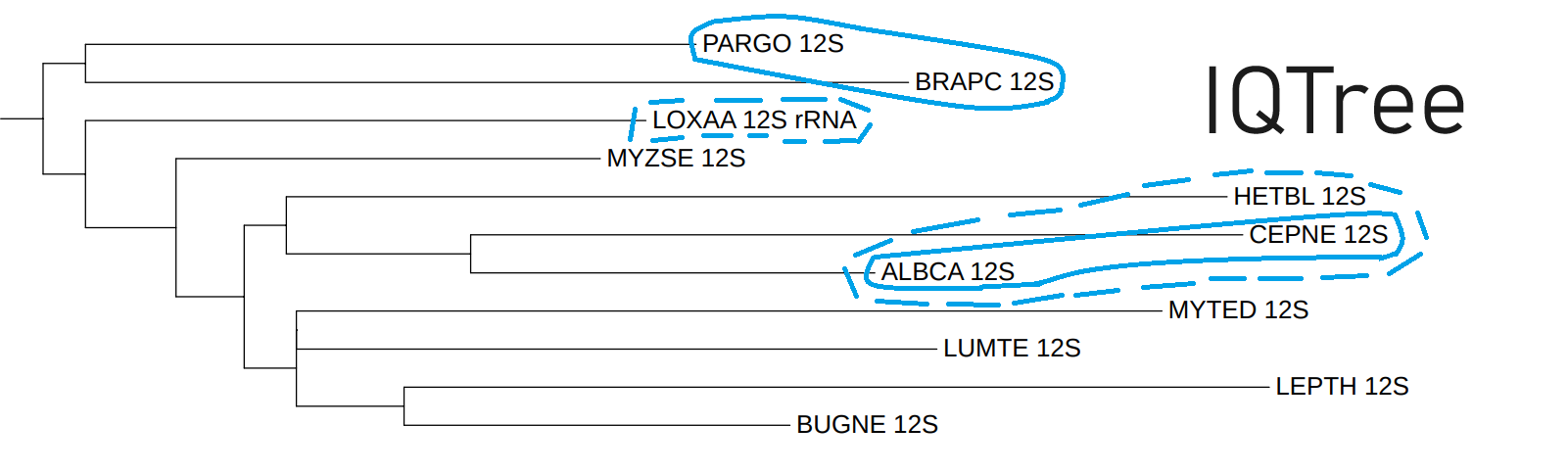
Это дерево не только правильно отделило базальную группу Gnathifera, включающую в себя BRAPC и PARGO, но и смогло правильно определить близкие виды одного рода Helicina: ALBCA и CEPNE. Из положительных черт это то, что дерево сблизило HETBL, CEPNE и ALBCA (принадлежат моллюскам) и выкинуло LOXAA за пределы, не приписав его ни к одной другой группе внутри Lophotrochozoa.
Укоренение во внешнюю группу
Сестринской к кладе Spiralia является группа Ecdysozoa, обе эти группы принадлежат к первичноротым Prostomia. В качестве внешней группы я решил сначала взять представителя отряда мечехвостов - Limulus polyphemus. Сначала я разберу деревья, построенные по аминокислотным последовательностм цитохрома b:

Каких-то кардинальных изменений не произошло:
правильные группы остались правильными, неправильные группы остались неправильными. Из положительных черт можно выделить то, что в дереве с внешней группой организм с мнемоникой LOXAA не сближен ни с одним другим организмом (так и должно быть).
Дерево по нуклеотидной последовательности 12S рРНК, полученное при помощи IQtree получилось неплохим. И мне стало интересно, станет ли оно лучше, если добавить внешнюю группу (спойлер: нет):
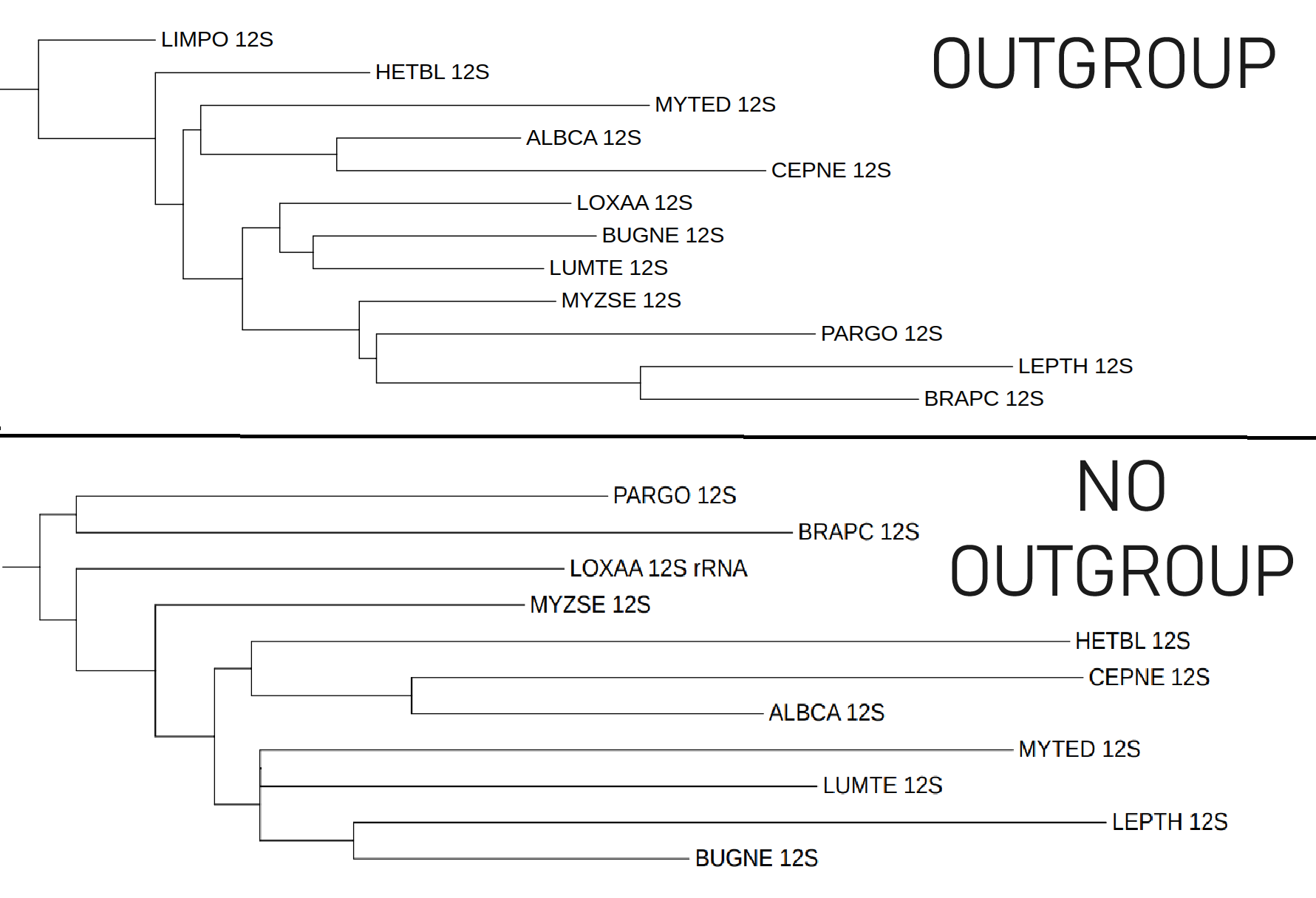
По неизвестным причинам дерево с внешней группой намного хуже отражает реальность, чем дерево без внешней группы. Это видно хотя бы по тому, что:
К Gnathifera (BRAPC+PARGO) каким-то образом занесло LEPTH (принадлежащего Lophotrochozoa), HETBL - отделился от моллюсков (CEPNE+ALBCA+MYTED+HETBL), Единственный плюс дерева с внешней группой – это то, что MYTED стал сближен с другими представителями моллюсков ALBCA и CEPNE.
Бутстреп
Дерево на рисунке 4, построенное алгоритмом IQtree по нуклеотидным последовательностям малой митохондриальной рРНК (=12S рРНК) оказалось самым лучшим (относительно всех остальных плохих деревьев). И для того, чтобы проверить, является ли данный результат случайностью или программа действительно смогла разобраться с филогенией Spiralia, я намерен использовать бутстрэп. Для этого я использовал команду:
iqtree -s 12S.phy -b 100 -redo
"-b 100" – делает 100 реплик, "-redo" – позволяет повторно использовать команду IQtree на одном и том же файле с выравниваниями.
В итоге у меня получилось следующее дерево:
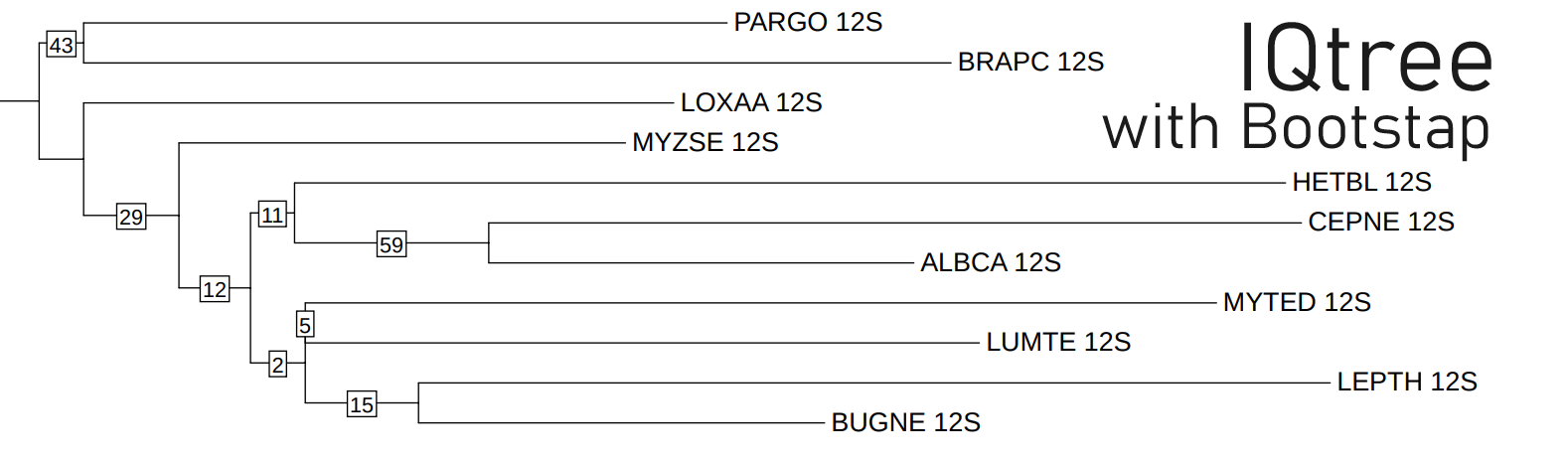
Из рисунка видно, что правильные (с таксономической точки зрения) группы, действительно, имеют неплохую восппроизводимость (43 у Gnathifera – PARGO+BRAPC; 59 у Helicina - CEPNE+ALBCA). Неправильные же группы LEPTH+BUGNE или LUMTE+MYTED имеют низкую воспроизводимость (значение бутстрепа) – 15 и 5 соответственно. Однако сближения HETBL (представителя моллюсков) с ALBCA+CEPNE (также представителями моллюсков) имеет низкую вопроизводимость - 11, хотя с точки зрения таксономии – это правильный ход. Интересно то, что группа из (MYZSE+HETBL+CEPNE+ALBCA+MYTED+LUMTE+LEPTH+BUGNE) выделяется с неплохой воспроизводимость в 29, это означает, что в 29 из 43 случаев, когда выделяются Lophotrochozoa (LOXAA+MYZSE+HETBL+CEPNE+ALBCA+MYTED+LUMTE+LEPTH+BUGNE) организм LOXAA будет отходить раньше всех, что в принципе не так далеко от истины (это можно понять, если посмотреть на таксономические дерево)