Практикум 8. Сигналы и мотивы
Описание мотива в белках паттерном
В качестве мнемоники белка я выбрал FADJ. Белки данного типа являются альфа-субъединицей комплекса окисления жирных кислот. Они катализируют реакцию образования гидроксиацил-КоА путем присоединения воды к эноил-Коа.

По базе данных Swiss-Prot при помощи запроса среди бактерий:
(id:FADJ_*) AND (taxonomy_id:2)
было найдено 78 белков с мнемоникой FADJ
Я решил взять 10 из них:
FADJ_ALIF1
FADJ_CITK8
FADJ_CROS8
FADJ_ECOLI
FADJ_PHOPR
FADJ_SALTY
FADJ_SHEON
FADJ_SHIF8
FADJ_VIBCH
FADJ_YERPE
Скачал их белковые последовательности в формате Fasta, затем выровнял их программой Muscle, получив следующее выравнивание
Я нашел очень интересный консервативный гидрофобный участок с большим количество глицина:
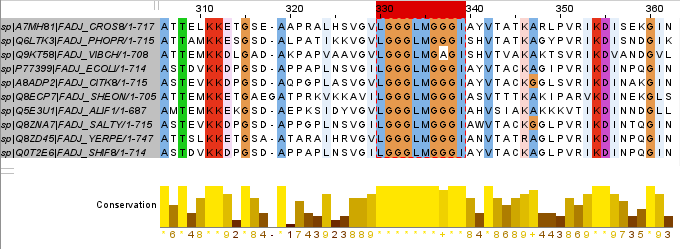
Белки длиной около 700 аминокислот, а начальная позиция мотива чаще 314-ая аминок-та, реже 316-ая аминок-та.
По этому выравниванию можно составить следующий паттерн:
L-G(3)-L-M-G-[GA]-G-I
Затем по этому паттерну я осуществил поиск белков с мнемоникой FADJ среди всех бактериальных белков из Swiss-Prot при помощи программы fuzzpro:
fuzzpro -sequence "/P/y24/term4/bacteria-sw.fasta" -pattern "L-G(3)-L-M-G-[GA]-G-I" -outfile "fadj_report.fuzzpro"
Я получил следующий выходной файл.
Среди всех 336944 белков программа fuzzpro по паттерну смогла найти 78 белков, среди которых оказались все нужные белки с мнемоникой FADJ. То есть по этому паттерну не было ни ложноположительных, ни ложноотрицатльных находок.
Так как паттерн сработал идеально, не думаю, что он требует улучшений
Поиск мотивов в белках программой MEME и поиск этих мотивов в банке
Для этих белков я также решил найти мотивы, используя программу MEME:
meme FADJ.fasta -protein -minw 8 -maxw 15 -mod oops -oc fadj_meme_out -nmotifs 3
'-protein' – аминокислотные последовтельности
'-minw 8' и '-maxw 15' – найти мотив длиной от 8 до 15 аминокислот
'-mod oops' – в каждой последовательности мотив встречается один раз
'-nmotifs 3' – найти до трех мотивов в этих белках
Найденные мотивы можно увидеть в выходном html-файле:
Они все максимально допустимой длины в 15 аминокислот (Width=15). Присутствуют во всех аминокислотных последовательностях (Sites=10). Имеют очень высокую значимость (E-value<10^-93 для всех находок). В данном случае E-value означает матожидание числа находок мотивов с таким же или большим информационном содержании в таком же числе последовательностей таких же длин.
Данный html-файл я подал в программу MAST, которая будет искать данные мотивы в бактериальных белках из Swiss-Prot:
mast 'fadj_meme_out/meme.html' '/P/y24/term4/bacteria-sw.fasta' -oc fadj_mast_out
Я получил следующий выходной html-файл. Всего нашлось 230 последовательностей, чьё E-value меньше 10. В данном контексте E-value означает математическое ожидание находок белков в случайной базе данных того же размера, которые совпали с мотивами так же хорошо, как и данная последовательность. E-value здесь высчитывается по схожести сразу со всеми поданными на вход мотивами.
78 находок с самым низким E-value (<2.5e-35) являются белками с мнемоникой FADJ. Самый низкий E-value для белка с мнемоникой, отличной от FADJ, равен 2.4e-19. Поэтому, если мы хотим среди находок получить только белки с мнемоникой FADJ мы можем сделать фильтрацию по E-value:
mast 'fadj_meme_out/meme.html' '/P/y24/term4/bacteria-sw.fasta' -oc fadj_mast_restricted_out -ev 1e-19
Помимо белков с мнемоникой FADJ, программа MAST нашла сходство с входными мотивами у белков со следующими мнемониками: FADB, HBD, FADB2, LCDH, PAAH
FADB - также является альфа-субъединицей коплекса окисления жирных кислот, однако в отличие от FADJ принимает активное участие не только в анаэробном разложении жирных кислот, но и в аэробном.
HBD - проявляет ту же 3-гидроксилбутирил КоА-дегидрогеназную активность, что и FADJ, но только по отношению к короткоцепочечным жирным кислота – бутирату
FADB2 - также обладает 3-гидроксилбутирил КоА-дегидрогеназной активностью
LCDH - также является дегидрогеназой, но окисляет уже не жирные кислоты, а L-карнитин
PAAH - тоже проявляет активность 3-гидроксибутирил-КоА-дегидрогеназы
Становится очевидно, что мотивы, полученные программой MEME играют важную роль 3-гидроксиацил-КоА-дегидратазной активности белка.
Поиск последовательности Шайна-Дальгарно в геноме Thermus brockianus
Геном бактерии Thermus brockianus состоит из 3-ёх молекул: хромосомы, мегаплазмиды и плазмиды. Я буду анализировать все три последовательности.
Чтобы найти последовательность Шайна-Дальгарно в геноме, нужно сначала создать паттерн; для большинства бактерий ПШД имеет следующий вид: AGGAGG. Чтобы найти все такие паттерны в молекулах я использовал следующую команду:
fuzznuc -sequence 'thermus_brockianus.fna' -pattern 'A-G-G-A-G-G' -complement -outfile 'direct_and_complement_shine_dalgarno.fuzznuc'
Она будет искать вхождения как на плюс цепи (прямой), так и на минус цепи (обратной комплементарной). При выполнении она выдала следующий файл.
Из него видно, что в трёх последовательностях программа смогла найти 13359 совпадений с паттерном последовательности Шайна-Дальгарно. Однако сложно сказать, много ли это или мало, поэтому посчитаем ожидаемое число находок.
С этой целью я использовал следующий код на Python. Он позволяет найти обогащение определенного паттерна, то есть во сколько раз реальное число находок больше (или меньше) ожидаемого числа находок. Сначала я решил посчитать ожидаемое число находок, используя частоты мононуклеотидов в последовательностях:
| Цепь | Хромосома | Мегаплазмида | Плазмида |
|---|---|---|---|
| + | 8.19 | 7.12 | 5.30 |
| - | 8.22 | 7.38 | 5.55 |
Однако эти цифры мало, что говорят о реальном обогащении ПШД. Вдруг подслова в последовательности Шайна-Дальгарно имеют повышенную частоту встречаемости, вследствие чего повышена частота находок ПШД. Чтобы это опровергнуть, я использовал формулу для учета частоты подслов[1].
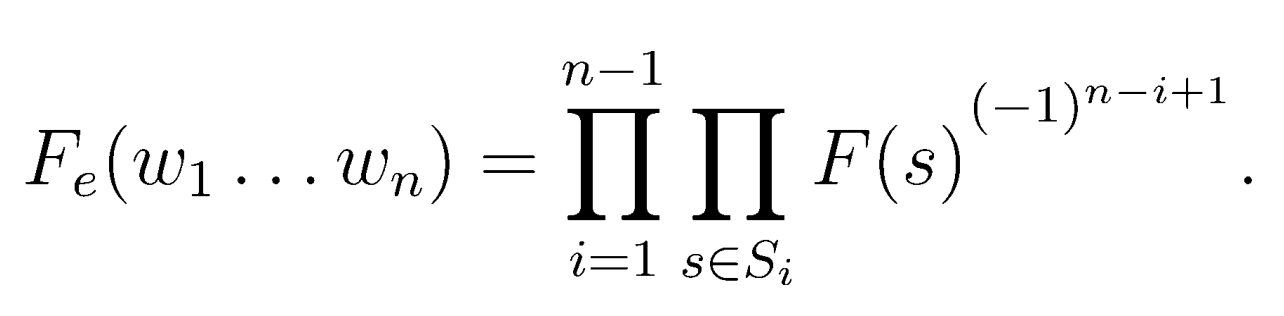
В результате я получил следующие цифры обогащения ПШД в геноме Thermus brockianus для разных цепей:
| Цепь | Хромосома | Мегаплазмида | Плазмида |
|---|---|---|---|
| + | 2.31 | 2.07 | 1.89 |
| - | 2.33 | 2.05 | 1.82 |
Данные цифры обогащения нам уже говорят о многом: AGGAGG - встречается примерно в два раза чаще в геноме, чем мы бы этого ожидали. При этом в геномной хромосоме последовательность Шайна-Дальгарно встречается чаще, чем в плазмидах. При этом ПШД встречается чаще именно в мегаплазмиде.
Скорее всего это объясняется тем, что в геномной хромосоме и мегаплазмиде сосредоточены жизненно важные гены, в то время, как в маленькой плазмиде находятся вспомогательные гены. Вследствие чего последняя более подвержена мутагенезу и последовательности Шайна-Дальгарно чаще исчезают.
Последовательность Шайна-Дальгарно - это последовательность перед старт-кодоном (около 10 нуклеотидов выше по цепи), которую узнаёт 16S рРНК маленькой субъединицы рибосомы.
Мы хотим найти именно последовательность Шайна-Дальгарно, а не случайную AGGAGG, для этого мы будем проверять каждую находку на то, действительно ли она лежит перед старт-кодоном.
Для этого я использовал следующую последовательность команд:
awk '/Sequence:/ {seq=$3} $3=="+" {print seq"\t"$1"\t"$2}' complement_and_direct_shine_dalgarno.fuzznuc > AGGAGG_plus.tsv
awk '/Sequence:/ {seq=$3} $3=="-" {print seq"\t"$1"\t"$2}' complement_and_direct_shine_dalgarno.fuzznuc > AGGAGG_minus.tsv
Если находит в файле строку, содержащую 'Sequence:', то присваивает seq название последовательности (слово после Sequence);
Выводит через табуляцию: название последовательности, координату начала находки ПШД, координату конца находки ПШД
grep 'CDS' themus_brockianus.gff > thermus_brockianus_CDS.gff
Из таблицы особенностей извлекает только кодирующие последовательности
cut -f 1,4,7 thermus_brockianus_CDS.gff | grep '+' | cut -f 1,2 > plus_chain.tsv
Извлекает из кодирующих последовательностей название цепи и координату начала старт-кодона гена (самое маленькое число) на плюс цепи
cut -f 1,5,7 thermus_brockianus_CDS.gff | grep '-' | cut -f 1,2 > minus_chain.tsv
Извлекает из кодирующих последовательностей название цепи и координату начала старт-кодона гена (самое большое число) на минус цепи
awk 'NR==FNR {cds[$1][$2]; next} {status = "FALSE"; if ($1 in cds) {for (pos in cds[$1]) {diff=pos-$3; if (diff>0 && diff<=20) {status="TRUE"; break} } } print $0 "\t" status}' plus_chain.tsv AGGAGG_plus.tsv > AGGAGG_plus_marked.tsv
NR - счетчик строк с начала запуска, FNR - счетчик строк в текущем файле. Условие NR==FNR выполняется только при чтении первого файла. Мы для каждой последовательности записываем координаты начала гена.
Когда условие NR==FNR не выполняется, то есть мы читаем второй файл, проверяем каждую находку на то, найдется ли ген, перед которым лежит находка в пределах 20 нуклеотидов.
К текущей строке файла с находкамис мы дописываем статус: находится ли данная находка перед начало гена
awk 'NR==FNR {cds[$1][$2]; next} {status = "FALSE"; if ($1 in cds) {for (pos in cds[$1]) {diff=$2-pos; if (diff>0 && diff<=20) {status="TRUE"; break} } } print $0 "\t" status}' minus_chain.tsv AGGAGG_minus.tsv > AGGAGG_minus_marked.tsv
Аналогично для минус цепи
Если подсчитать, количество находок, у которых status=TRUE, то есть находки, находящиеся в пределах 20 нуклеотидов перед старт-кодоном, то для обоих цепей их получится около 460 из 13359 (3.44%)
Если проводить грубые расчёты, то на трёх последовательностях на обеих цепях у нас суммарно: 2.4*2=4.8 млн пар оснований и 2500 генов кодирующих белки. Для каждого гена мы берём 20 нуклеотидов, то есть суммарно 2500*20=50 000 нуклеотидов, тогда примерное и грубое матожидание числа находок перед генами составляет 13 359*50 000/4 800 000 = 139.2 штуки; реальное число находок в 460/139=3.3 раза больше, чем ожидаемое число находок. Эти грубые расчёты позволяют нам прийти к выводам, что число 460 неслучайно и гораздо выше ожидаемого.
Ссылки на статьи