{kind=link}

 Back to the second term
Back to the second term
Для выполнения поставленной задачи были приготовлены три файла с последовательностями в формате fasta:
myprot.fasta — Последовательность фосфоенолпируваткарбоксилазы кишечной палочки (CAPP_ECOLI).
secondprot.fasta — Последоваетльность гомолога нашего белка (CAPP_ECOLI) CAPP_YERPE из Yersinia Pestis.
thirdprot.fasta — Искусственно созданная (посредством копирования и вставления CCCV)последовательность из двух небольших (10–12 букв каждый) участков аминокислотной последовательности белка CAPP_ECOLI.
Чтобы проделать такое выравнивание, использовались командная строка Unix и две, строющие выравнивания программы: Needle и Water, первая из которых выстраивает, согласно алгоритму Нидельмана-Вунша,глобальное выравнивание двух последовательностей, вторая же также строит выравнивание последовательностей,однако уже локальное и по принципу алгоритма Смита-Ватермана. Узнать подробнее о вышеназванных алгоритмах можно пройдя по ссылке на страницу "Матрицы переходов глобального и локального выравнивания".
Штраф за открытие гэпов (Gap opening penalty) 10.0, за продление гэпов (Gap extension penalty) 0.5,
параметры взяты по умолчанию, согласно стандартным настройкам программ. Использовалась матрица
аминокислотных замен типа BLOSUM62.
В итоге мы имеем два документа с глобальным и локальным выравниваниями:
Теперь попытаемся сравнить эти выравнивания друг с другом. Есть несколько стандартных характеристик, которыми определяется каждое выравнивания независимо, в какой программе и с помощью какого алгоритма оно сделано. Во-первых, это вес выравнивания (Score). Вес вычисляется стандартным способом исходя из заданных параметров цены совпадения, открытия гэпов, их продолжения, а также аминокислотной замены. Во-вторых, процент идентичности (Identity). Эта характеристика вычисляется как отношение полностью совпавших аминокислотных остатков к общему числу аминокислот в максимально длинной из двух последовательности. И наконец, третий параметр — это процент схожести (Similarity). Он вычисляется так же, как и процент идентичности с той только разницей, что в числителе к совпавшим аминокислотам прибавляются те аминокислотные остатки, которые не совпали, однако чрезвычайно близки по своим физико-химическим свойствам (например, лейцин и изолейцин), что даёт нам право принимать такое "аминокислотное столкновение" за совпадение,хотя цена такого совпадения по идее должна быть поменьше непосредственно "чистого" совпадения.
Такие результаты говорят о том, что и глобальное, и локальное выравнивания абсолютно повторили друг друга и представляют собой одно и то же выравнивание. Почему так? Казалось бы, глобальное и локальное выравнивания довольно разные вещи и алгоритмы также неидентичны по идее. Однако такое, видимо, может случиться в связи с высокой гомологичностью двух последовательностей. Ведь стоит только посмотреть на значение веса выравниваний как становится "не по себе" — слишком велик вес, процент идентичности также достаточно высок, однако, лишив себя права на ошибку ещё на предыдущей странице, мы заключили абсолютно однозначно, что фосфоенолкарбоксилаза E.coli и изучаемый на гомологичность белок CAPP_YERPE действительно являются гомологами, "близкими родственниками", если конечно, так можно выразиться. Итак, две принципиально разные программы дали нам два идентичных выравнивания. Вывод напрашивается сам собой — существует только одно наилучшее выравнивание, которое воспроизводится путём обоих алгоритмов, которые кстати, довольно близки по своим принципам, что тоже может являться дополнительным объяснением такого "тотального" совпадения двух выравнивания, глобального и локального.
Для выполнения этой задачи снова было предложено поработать с двумя уже известными нам прграммами Needle и Water, а также с ещё одной похожей программой Matcher, тоже строющей, как и Water, локальные выравнивания, однако позволяющей в одном файле-результате с выравниванием построить не одно единственное лучшее выравнивание, а несколько, при этом число выравниваний в одном файле можно задать произвольное, согласно поставленным требованиям. В нашем случае это число равно трём.
Штраф за открытие гэпов (Gap opening penalty) 10.0, за продление гэпов (Gap extension penalty) 0.5, параметры взяты по умолчанию, согласно стандартным настройкам программ. Использовалась матрица аминокислотных замен типа BLOSUM62.
Начнём с самого первого выравнивания. Сразу, что бросается в глаза — очень низкие проценты идентичности, схожести, гэпов. Но это и неудивительно и объясняется огромной разностью в длинах обоих последовательностей. На этом примере отчётливо видно, как собственно работает Needle: В первую очередь ищется самый большой и длинный участок с совпадающими аминокислотами. К такому заключению приводит тот факт, что программа выровнила последовательность по более длинному куску ADLWLAEYYDQRL. Второй кусок, более короткий, выравнивается постольку поскольку, то есть программа, даже если и нашла идентичный кусок в последовательности белка, то просто проигнорировала его, дабы не открывать космическое количество гэпов. Плавно переходя ко второму файлу мы видим, что локальное выравнивание построено также на более длинном куске ADLWLAEYYDQRL, все остальные гэпы и второй кусок QVIAEAPQGSIAA просто выброшены. Отсюда понятно, как действует Water: словно Needle, ищет наиболее длинный участок совпадающих аминокислотных остатков, а затем отбрасывает всё ненужное в виде оставшейся части последовательности и огромного числа гэпов, которые сохранялись в Needle в связи с тем, что строилось не локальное, а глобальное выравнивание.
Теперь что касается последней программы. Matcher выдала нам три выравнивания, первые два из которых являются стопроцентноидентичными теми самыми участками последовательности белка, из которых мы моделировали маленькую вторую последовательность. На первом месте опять же самое ценное выравнивания с более длинным куском ADLWLAEYYDQRL, затем выравнивание по QVIAEAPQGSIAA. Третье выравнивание представляет собой случайно совпавший набор аминокилот, которые на самом деле можно назвать "шумом", потому что проистекают из чистой случайности. Просто в последовательности белка, которая примерно в 35 раз длиннее искусственной, нашлись похожие участки. Третье выравнивание имеет небольшой вес по сравнению с двумя первыми, но поскольку параметр числа выравниваний выставлет равный трём, программе, так сказать, некуда деваться и нужно выдать именно три выравнивания, тогда как мы знаем, что на самом деле правильных выравниваний существует только два. На третье выравнивание обращать внимание не стоит, оно иллюстрирует только то, что делает программа после того, как нашла все возможные правильные выравнивания, а ей ещё нужно, согласно выставленным требованиям, построить n-ое количество выравниваний. Стоит пояснить, что программа не в состоянии определить, какое выравнивание является правильным, а какое относится к "шуму", она, как правило, руководствуется только величиной веса и заданными штрафами за открытие и продолжение гэпов.
Теперь сравним три глобальных выравнивания тех же последовательностей, что и в предыдущем пункте, сделанные прогрммой Needle, построенных при различных значениях такого параметра, как штарф за открытие гэпов. Штраф за продолжение гэпов мы не меняем и положим равным единице.
| Штраф за открытие гэпа | Штраф за продолжение гэпа | Файл результатов |
| 10 | 1 | 1to3_10_1.needle |
| 5 | 1 | 1to3_5_1.needle |
| 1 | 1 | 1to3_1_1.needle |
Что касается первых двух выравниваний, то они можно сказать одни и те же, если не брать во внимание разное значение весов.Говоря "одни и те же", я не ни в коем случае не имею в виду то, что они являются идентичными, это означает лишь то что, аминокислоты расположены одинаково друг относительно друга. Вес больше естественно там, где меньше значение штрафа за открытие гэпов, то есть при его значении равном 1. Третье выравнивание выбивается из общей картины. Поскольку штраф теперь минимален, программа уже не "боится" открыть лишние гэпы, поэтому она разбивает тот участок маленькой последовательности так, чтобы получить максимальное число совпадений и полусовпадений., таким образом число гэпов повышается, однако вес выравнивания в любом случае остаётся большим, нежели в первых двух случаях, это связано опять же с меньшим штрафом за открытие гэпов.
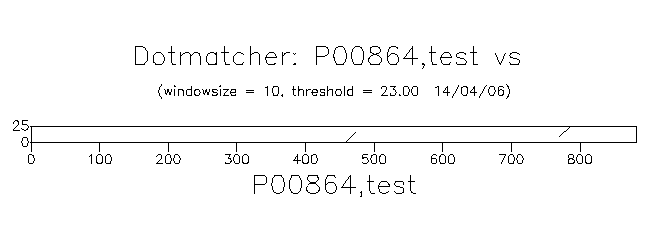
С помощью программы dotmatcher мы построили карту локального сходства последовательностей из myprot.fasta и thirdprot.fasta. .У нас есть параметры, которые необходимо изменять последовательно, дабы понять как подобные измения влияют на вид карты локального сходства и что собственно они значат. Это Threshold и Windowsize. Параметр типа используемой матрицы (Matrix type) не меняется, во всех случаях используется матрица EBLOSUM62.
Стандартное значение параметра - 23. Не меняя второй параметр размер окна, мы построили карты локального сходства с порогами на суммарный вес соответственно равными 7, 23 (стандарт), 30, 45 и 75. На картах можно увидеть некие полоски, которые думается, соответствуют локальным выравниваниям. При значении 23 можно наблюдать всего две полоски, при значении 7 количество их рузко увеличивается, но при этом также резко уменьшается их длина — выравнивания становятся меньше. По мере увеличения веса у нас остаётся две полоски, до тех пор пока мы не достигнем некого минимума, при котором мы уже не увидим ни одного выравнивания. Этот минимум, несомненно, должен быть больше максимальнго веса среди всех выравниваний. Самый большой вес в нашем случае равен 72, что очевидно из файла 1to3.matcher, поэтому как только мы задаём порог на суммарный вес больше 72, полоски исчезают. Это хорошо видно на приведённом ниже рисунке.
Стандартное значение параметра - 10. Мы изменяем его, соответственно, таким образом: 3 (минимальное значение, допускаемое программой), 5, 10, 15, 20. Нетрудно заметить, что параметр размер окна влияет на длину полосок выравниваний. А что такое длина выравнивания? Пожалуй, это количество аминокислотных остатков, входящих в это выравнивания. Такое утверждение даёт нам повод заключить, что windowsize отвечает за степень допустимого сходства аминокислот, воспринимаемого программой. Чем больше параметр размера окна, тем больше аминокислотных пар программа примет за аминокислоты, входящие в выравнивание. Чем меньше размер окна, тем ниже степень допустимого сходства и даже если само реальное выравнивание окажется за пределами значения этого параметра, программа отрежет его несмотря ни на что - так получилось с минимальным значением windowsize - при его значении равном трём на карте мы не видим ни одного выравнивания, тогда как прекрасно знаем, что они существуют.

© Анна Чебышева,2005