Рис.1 Дерево, полученное с помощью drawtree 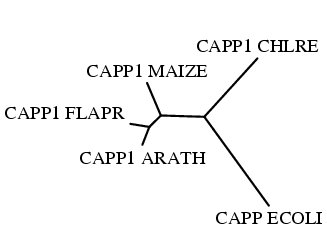
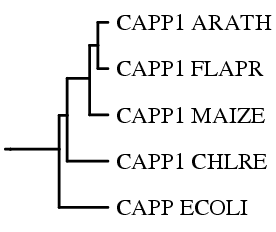
Рис.2 Дерево, полученное с помощью drawgram
Филогенетические деревья, реконструированные разными способами
На этот раз нам предложено попробовать изучить эволюционные взаимоотношения нескольких белков, руководствуясь данными множественного выравнивания их последовательностей, а точнее их общего домена. В качестве материала возьмём пять фосфоенолпируват карбоксилаз с общим доменом PEPCase. А именно:
Множественное выравнивание для них сохранено в файле PEPCase.msf С помощью редактора GenDoc получим матрицу попарной идентичности, представляющую собой сводку данных об идентичности всевозможных пар последовательностей пяти белков. Затем обратимся к предложенному ульраметрическому алгоритму построения филогенетических деревьев UPGMA (Unweighted Pair Group Method with Arithmetic Mean).
Построение дерева по алгоритму UPGMA
Создание Excel-книги
UPGMA.xls
На основе матрицы попарной идентичности, импортированной в Excel, была построена вторая матрица "наивных" эволюционных расстояний между последовательностями выравнивания. В основу положено предположение, что по мере возрастания процента идентичности сокращается эволюционное расстояние пары последовательностей. Исходя из общих соображений и элементарной логики это гипотетически верно. Договоримся считать эволюционным расстоянием (D) величину D = 100 – P, где P — процент идентичности. Посмотреть на обе матрицы можно на листе Excel-книги "Distances". Однако не стоит забывать, почему мы назвали эти расстояния "наивными". Потому что вычисляются они чрезвычайно грубо и не содержат большого объёма входных данных, ибо в нашем случае ими является всего лишь матрица попарной идентичности. Очевидно, что в процессе эволюции было задействовано достаточно много факторов, влияющих на её ход, так что нельзя с уверенностью полагаться на данные, полученные только на основе одного из них.
Построение дерева по алгоритму UPGMA:
((((CAPP1_ARATH:0.065,CAPP1_FLAPR:0.065):0.055,CAPP1_MAIZE:0.12):0.1475, CAPP1_CHLRE:0.2675):0.0525,CAPP_ECOLI:0.32);
На данном этапе дерево уже построено. Есть узлы, корни, листья, рёбра и их длины. Скобочная структура позволяет нам визуализировать полученное дерево с помощью таких программ, как drawtree и drawgram из пакета Phylip. Эти программы на основе скобочной структуры "рисуют" филогенетические деревья. Принципиальным в структуре являются расстояния, то есть цифры, которые в совокупности представляют собой соотношение, на основе которого программы "рисуют" рёбра. То есть если мы умножим все числа в скобочной структуре на миллион, то длины рёбер и само дерево не изменятся.
|
Рис.1 Дерево, полученное с помощью drawtree |
|
Рис.2 Дерево, полученное с помощью drawgram
|
Программа drawtree строит неукоренённое (Рис.1) филогенетическое дерево, а drawgram - укоренённое. Как одно из условий ультраметрического алгоритма UPGMA соблюдено одинаковое расстояние между узлом и листьями в пределах одного кластера. Оба дерева, как укоренённое, так и неукоренённое, отражают в целом одно и то же, ибо были построены на основе одной и той же скобочной структуры. То есть глядя на оба дерева мы можем точно сказать, что CAPP_ECOLI наиболее эволюционно отдалённый белок от всех остальных; можем сказать, что самая высокая степень родства наблюдается у пары белков CAPP1_FLAPR и CAPP1_ARATH и т.д.
С точки зрения различий тут сразу бросается в глаза общий принцип построения деревьев, заложенный в программах. В случае неукоренённого дерева (Рис.1) из схемы не следует существание общего гипотетического предка для всех пяти белков, что неверно для укоренённого дерева (Рис.2), глядя на которое мы можем сказать, что "да, эти белки несомненно произошли от одного предка.." В первом случае нет никаких намёков на то, как шла эволюцию, в какую сторону она шла и что в итоге получилось. Можно только догадываться, кто от кого произошёл и кто быстрее отщепился. Максимально, на что может ответить неукоренённое дерево, это как распределяется степень родства между белками. То есть легко заметить, что например CAPP1_FLAPR и CAPP1_ARATH, возможно, скорей всего, относятся к одной эволюционной линии в пределах данного семейства белков, и может быть, они произошли от одного предка.Данный тип визуализации, на мой взгляд хорош, в тех случаях, когда нет чётких представлений об эволюционных связях в пределах выборки белков. Что касается укоренённого дерева (Рис.2), то оно даёт нам ответ на все те, вопросы, кои остались без ответа первоначально. Как идёт эволюция? Смотрим, ага, гипотетический предок А даёт нам в процессе эволюции белок CAPP_ECOLI и некий белок В, послуживший предком для CAPP1_CHLRE и белка С, в свою очередь давшего начало белку CAPP1_MAIZE и белку D, от которого произошли теоретически CAPP1_FLAPR и CAPP1_ARATH. В каком направлении идёт эволюция? Очевидно, CAPP_ECOLI отщепился раньше, к примеру, чем CAPP1_CHLRE, то есть он древнее и соответственно появился на более ранних этапах эволюции, так что куда идёт эволюция, согласно укоренённому дереву, мы скажем в любом случае.
В целом неукорененное показывает "неродство" (потому что расстояние наглядно - насколько далеко одно от другого). А укорененное - пытается объединить в группы (кластерный анализ) и показывает, как могла проходить эволюция групп.
На первый взгляд, кажется, что случай второй визуализации укоренённого дерева более объемлющ по информации, более чёток и нагляден, однозначен и систематизирован. Да, конечно, на первый взгляд оно так. Но прежде чем отдавать лавры первенства укоренённым деревьям стоит задуматься, а верна ли в корне вся та информация, которой изобилуют эти деревья? Действительно, откуда знать дереву на самом деле, был общий предок у CAPP1_FLAPR и CAPP1_ARATH или не было, был ли вообще в теории какой-либо общий предок у всех пяти белков или CAPP_ECOLI и все остальные вообще две параллельные линии эволюции (хотя это конечно вряд ли, скорей всего существует n-ое количество разветвлений, прежде чем можно будет объеденить CAPP_ECOLI и всех остальных в одну линию эволюции). Да мы и без всяких деревьев можем только предполагать и строить догадки на основании таксономии, фунцкий, последовательностей, доменов, сайтов и пр., пр. об истинной эволюции этих белков, чтобы получить оную, необходимо огромное количество сведений о всех белках данного семейства, даже собрав их, вряд ли теория об эволюции наших белков будет истинной, так что дерево - это всего лишь достаточно грубая и недостоверная модель процесса эволюции, приближение, предположение, которое никак не может однозначно определять то, о чём оно предполагает, к чему приближается. Поэтому более адекватным и достоверным мне видится неукоренённое дерево. Ему и отдам своё предпочтение.
Последний вопрос о "кратком сценарии эволюции" видится мне не совсем уместным, ибо я ничего не могу сказать об эволюции этих белков, всё, что я знаю исходя из нукоренённого дерева, это:
Теперь попробуем построить филогенетические деревья не вручную через матрицу попарной идентичности, а с помощью
программного инструментария, коим мы уже обладаем. Сразу можно вспомнить программу ClustalW,
которая не только умеет строить множественные выранивания, но и генерирует наряду с ними и правильную скобочную структуру для построения
генерального дерева в визуализаторе. Эта структура выдаётся программой на выходе в файле NJ.dnd. Файл можно не
открывать =), вот так она выглядит:
((CAPP1_ARATH:0.06409,CAPP1_FLAPR:0.06130):0.00569,(CAPP1_CHLRE:0.26031, (CAPP2_CHLRE:0.28576,CAPP_ECOLI:0.28276):0.03778):0.18162,CAPP2_MAIZE:0.07036);
Скобочная структура иная. Действительно, во-первых, программа посчитала расстояния более точно, видимо,
хотела посчитать с точностью до пятого знака, но не знала, что это значит, поэтому где-то получилось до пятого, а где-то до четвёртого.
Далее, что мы замечаем, ещё не приступая непосредственно к визуализации? Ну, безусловно то, что больше рёбра одинаковыми по длине
никак не будут.
|
Рис.3 Дерево, полученное с |
Рис.4 Дерево, полученное с |
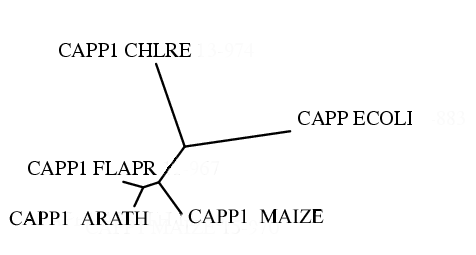
|
 |
Получив по новой скобочной структуре новые филогенетические деревья всё в тех же drawgram и drawtree, сравним результаты. В первом случае построения деревьев вручную, как уже было сказано, использовался ультраметрический алгоритм, согласно которому все длины рёбер одинаковы. Теперь же, судя по скобочной структуре, использовался явно алгоритм неультраметрический. Мне кажется, вполне разумно, что рёбра должны быть разной длины, ибо в то, что два белка отщепляются одновременно с течением времени и эволюции, мне слабо верится. Что касается UPGMA-неукоренённого и NJ-неукоренённого деревьев, то они практически идентичны. Последнее только слегка по-иному ориентировано, однако на общий смысл ориентация ребёр на плоскости не влияет, для нас важны узлы и длины рёбер. NJ-укоренённое дерево, как ни странно, отличается от UPGMA-укоренённого. CAPP_ECOLI и CAPP1_CHLRE объединяются в один кластер и полагается, что CAPP1_MAIZE - белок, произошедший на более ранних стадиях эволюции, чем CAPP1_ECOLI. Но с тривиальной точки зрения таксономии это невозможно, ибо говоря грубо, кукуруза никак не могла эволюционно опередить кишечную палочку. Поэтому данное дерево мне видится несколько странным.
В заключении, хочу отметить, что неукоренённое дерево, как UPGMA, так и
NJ, близки к действительности в большей степени, чем укоренённые. Даже построенные разными
алгоритмами деревья дают нам один и тот же результат, что может служить каким-никаким, но доказательством его достоверности.