Практикум #12
Данный практикум посвящен работе в BLAST (англ. Basic Local Alignment Search Tool – средство поиска основного локального выравнивания) — семейство компьютерных программ, служащих для поиска гомологов белков или нуклеиновых кислот, для которых известна первичная структура (последовательность) или её фрагмент. Используя BLAST, исследователь может сравнить имеющуюся у него последовательность с последовательностями из базы данных и найти последовательности предполагаемых гомологов. Программы BLAST:
-
blastp
медленное сравнение аминокислотной последовательности с целью поиска всех сходных аминокислотных последовательностей; -
blastn
медленное сравнение нуклеотидной последовательности с целью поиска всех сходных нуклеотидных последовательностей; -
blastx
переводит изучаемую нуклеотидную последовательность в кодируемые аминокислоты, а затем сравнивает её с имеющейся базой данных аминокислотных последовательностей белков; -
tblastn
изучаемая аминокислотная последовательность сравнивается с транслированными последовательностями базы данных секвенированных нуклеиновых кислот; -
tblastx
переводит изучаемую нуклеотидную последовательность в аминокислотную, а затем сравнивает её с транслированными последовательностями базы данных секвенированных нуклеиновых кислот.
Задание 1. Поиск гомологов белка (2Z,6E)-хедикариолсинтазы
Что было сделано?
- Написали описание параметров, включая Algorithm parameters, которые использовали при запуске сервиса BLAST;
- Сохранили полную таблицу находок;
- Построли множественное выравнивание выборки из 5 — 10 последовательностей из начала, середины и конца списка Description (результатов поиска BLAST);
- Отредактировали выравнивание, оставив в нем только гомологичные, по нашему мнению, белки;
- Обосновали гомологичность наших находок.
Описание параметров программы blastp
Ниже, на рисунке 1, изображена страница вводных параметров для программы blastp из семейства компьютерных программ BLAST. Список обозначений:
-
#0
выбор программы, с которой Вы будете работать. Функции этих программ кратко описаны выше.
-
#1
в текстовое поле можно ввести или аминокислотную последовательность белка, или его AC, или его ID (а ниже, в поле Or, upload file можно также загрузить файл с последовательностью); -
#2
учитывать в поиске только те аминокислоты Вашей последовательности, от какой по какую указано в двух текстовых полях (по умолчанию учитываются все); -
#3
в текстовое поле можно ввести название последовательности в данном поиске; -
#4
можно выставить флажок, чтобы произвести поиск и выравнивание более чем двух последовательностей.
-
#5
из списка баз данных произвести выбор базы, в которой будет происходить работа; -
#6
произвести поиск только по указанным в текстовом поле организмам, опционально. Можно выставить флажок exclude, тогда будет происходить поиск только по тем организмам, которые не указаны в текстовом поле; -
#7
исключить из работы некоторые последовательности, опционально. Исключаемые последовательности можно выбрать, выставив нужные флажки.
Models (XM/XP): исключить последовательности, аннотации которых сделаны автоматически,
Non-redundant RefSeq proteins (WP): исключить референсные последовательности (исходники),
Uncultured/environmental sample sequences: исключить последовательности, полученные из организмов в естественной среде обитания (которые не были выращены в культуре);
-
#8
произвести выбор алгоритма поиска и выравнивания, выставив соответствующие флажки. Автоматически выставлен флажок blastp (protein-protein BLAST); -
#9
Начать бластование!!! Результаты можно получить в отдельной вкладке, выставив флажок.
-
#10
максимальное количество последовательностей, выводимое в результатах работы по данному запросу. Автоматически выбрано число 100; -
#11
автоматическое регулирование размера слова (это понятие связано с механизмом работы BLAST, смотрите выше по ссылке) для коротких последовательностей. Можно дезактивировать, выставив флажок. Он автоматически выставлен; -
#12
выставить в текстовом поле максимальное значение E-value (тоже связано с механизмом работы). Отображаются только те результаты, E-value которых меньше заданного. Чем меньше E-value, тем больше максимальный вес в битах, соответственно, тем более идентичные по отношению к исходному запросу будут результаты. Автоматически выбрано число 10; -
#13
выбрать из списка длину слова (связано с механизмом работы), на которые будет разбиваться заданная последовательность для отсортировки последовательностей из банка, подлежащих проведению среди них поиска. Автоматически выбрано максимальное число 6; -
#14
ввести в текстовое поле ограничение максимального количества совпадений между данной и искомой последовательностями. Необходимо в том случае, если в результатах содержится очень много сильно идентичных последовательностях, что может привести к тому, что BLAST не сможет вывести менее идентичные, но интересные результаты. Автоматически выставлено число 0, это значит, что никакого отбрасывания сильно идентичных последовательностей происходить не будет.
-
#15
выбрать из списка матрицу для вычисления очков выравнивания. По умолчанию - BLOSUM62; -
#16
выбрать из списка тип штрафов за гэпы (пропуски) в выравнивании; -
#17
выбрать из списка тип автоматического изменения (масштабирования) значений матрицы подстановок.
-
#18
исключить последовательности низкой сложности (например, аргинин-глицин богатые последовательности, повторы) которые могут привести к неочевидным и нелогичным результатам; -
#19
производить поиск нетривиальным способом.
Mask for lookup table only: производить выравнивания согласно сгенерированной хеш-таблицой поиска и только согласно с ней.
Mask lower case letters: производить фильтрование по строчным символам последовательности (скрыть строчные символы).
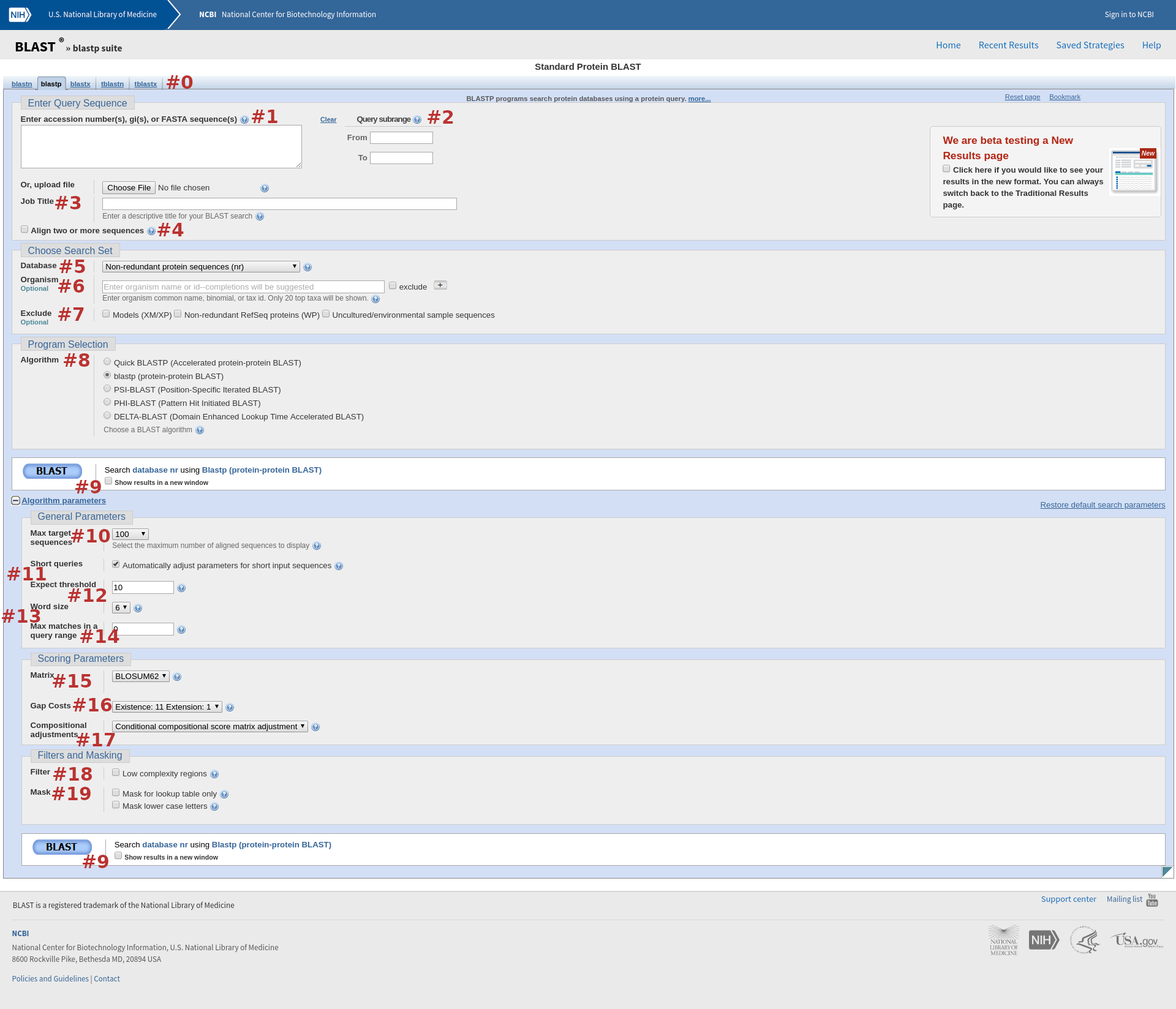
Рисунок 1.
Страница программы blastp на сайте BlastДополнительные материалы
Ниже распологаются изображения трех выравниваний для аминокислотных последовательностей, полученных из BLAST. Выравнивания сделаны в программе muscle, изображения получены через программу Jalview, раскраска — ClustalX. На каких именно последовательностях производилось выравнивание — описано ниже.

Рисунок 2.
Выравнивание белка HCS_KITSK с наиболее схожими последовательностями. Процент соответствия (identity) у них выше 70%, E-value меньше 10-164. Величина покрытия больше 95%.
Рисунок 3.
Выравнивание белка HCS_KITSK с последовательностями средней степени схожести. Процент соответствия (identity) у них выше 30%, E-value меньше 10-41. Величина покрытия больше 94%.
Рисунок 4.
Выравнивание белка HCS_KITSK с последовательностями малой степени схожести. Процент соответствия (identity) у них выше 25%, E-value меньше 10-18. Величина покрытия больше 90%.
Рисунок 5.
Распределение 100 лучших результатов запроса BLAST.Пояснение. Сверху, в секции Query seq. изображена линейная последовательность белка–запроса; на ней изображены очень специфические части последовательности, которые BLAST смог найти. Эти подпоследовательности относятся к доменам, которые изображены ниже у секции Superfamilies. Ниже изображены собственно результаты поиска. Здесь графически представлено покрытие каждого результата (мы его можем представить по длине и расположению относительно последовательности запроса). Также здесь представлены величены E-value, оценить которые мы можем с помощью цветов. Красным окрашены последовательности с очень низким E-value.
| # | Query (q) AC | Subject (s) AC | Identity, % | Alignment length | Mismatches | Gap opens | Q. start | Q. end | S. start | S. end | E–value | Bit score | Positives, % |
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| 0 | HCS_KITSK | WP_014133196.1 | 100.000 | 338 | 0 | 0 | 1 | 338 | 1 | 338 | 0 | 688 | 100.00 |
| 1 | HCS_KITSK | 4MC0_A | 100.000 | 338 | 0 | 0 | 1 | 338 | 1 | 338 | 0 | 687 | 100.00 |
| 2 | HCS_KITSK | RPE50111.1 | 72.531 | 324 | 89 | 0 | 1 | 324 | 1 | 324 | 1.04 * 10-167 | 480 | 81.17 |
| 3 | HCS_KITSK | WP_059414878.1 | 70.393 | 331 | 98 | 0 | 1 | 331 | 1 | 331 | 1.71 * 10-167 | 479 | 80.97 |
| 4 | HCS_KITSK | GAP46009.1 | 70.245 | 326 | 97 | 0 | 6 | 331 | 1 | 326 | 1.44 * 10-164 | 471 | 80.98 |
| 5 | HCS_KITSK | WP_086598900.1 | 68.580 | 331 | 104 | 0 | 1 | 331 | 1 | 331 | 5.66 * 10-160 | 460 | 79.15 |
| 6 | HCS_KITSK | WP_078916148.1 | 62.112 | 322 | 120 | 2 | 1 | 321 | 1 | 321 | 9.75 * 10-127 | 375 | 73.60 |
| 7 | HCS_KITSK | WP_115913110.1 | 61.801 | 322 | 121 | 2 | 1 | 321 | 1 | 321 | 5.12 * 10-126 | 374 | 73.29 |
| 8 | HCS_KITSK | KMS75306.1 | 61.356 | 295 | 112 | 2 | 28 | 321 | 1 | 294 | 3.93 * 10-109 | 330 | 73.22 |
| 9 | HCS_KITSK | WP_050375609.1 | 43.000 | 300 | 169 | 1 | 28 | 325 | 1 | 300 | 2.02 * 10-72 | 237 | 56.33 |
| 10 | HCS_KITSK | WP_093858087.1 | 35.294 | 323 | 203 | 3 | 4 | 321 | 2 | 323 | 2.07 * 10-47 | 172 | 51.39 |
| 11 | HCS_KITSK | WP_049714840.1 | 36.478 | 318 | 198 | 2 | 3 | 317 | 23 | 339 | 1.37 * 10-44 | 165 | 48.43 |
| 12 | HCS_KITSK | RPK77301.1 | 35.185 | 324 | 203 | 5 | 1 | 321 | 1 | 320 | 5.87 * 10-44 | 163 | 49.38 |
| 13 | HCS_KITSK | SCE18688.1 | 35.185 | 324 | 203 | 5 | 1 | 321 | 1 | 320 | 1.17 * 10-43 | 162 | 49.38 |
| 14 | HCS_KITSK | SFY17167.1 | 35.494 | 324 | 202 | 5 | 1 | 321 | 1 | 320 | 1.31 * 10-43 | 162 | 49.69 |
| 15 | HCS_KITSK | RAJ67521.1 | 33.742 | 326 | 206 | 5 | 1 | 321 | 1 | 321 | 9.27 * 10-42 | 157 | 50.31 |
| 16 | HCS_KITSK | ESU46049.1 | 33.746 | 323 | 209 | 4 | 1 | 321 | 1 | 320 | 1.20 * 10-41 | 157 | 49.23 |
| 17 | HCS_KITSK | WP_052687012.1 | 32.258 | 310 | 207 | 2 | 8 | 316 | 22 | 329 | 1.71 * 10-41 | 157 | 47.10 |
| 18 | HCS_KITSK | WP_053923023.1 | 34.307 | 274 | 177 | 2 | 8 | 280 | 11 | 282 | 1.76 * 10-41 | 157 | 50.00 |
| 19 | HCS_KITSK | SCF61559.1 | 33.746 | 323 | 209 | 4 | 1 | 321 | 1 | 320 | 2.15 * 10-41 | 157 | 48.92 |
| 20 | HCS_KITSK | SCF80729.1 | 33.746 | 323 | 209 | 4 | 1 | 321 | 1 | 320 | 4.37 * 10-41 | 156 | 48.61 |
Таблица 1.
Результат запроса BLAST, 21 первый результатПоиск гомологов
После загрузки аминокислотной последовательности белка HCS_KITSK
[E4MYY0 (2Z,6E)-hedycaryol
synthase {ECO:0000303 | PubMed:24399794} (HcS {ECO:0000303 | PubMed:24399794}) (4.2.3.187
{ECO:0000269 | PubMed:24399794})]
в текстовое поле Enter
FASTA sequence(s), смены максимального количества результатов на 20 000 (максимальное
возможное количество результатов, оно было выставлено для того, чтобы сравнить результаты с
разными
E-value) и нажатия на кнопку BLAST появилась веб-страница с результатами.
На ней мы можем увидеть многое. Например, изображение с распределением 100 лучших
результатов запроса BLAST (рисунок 5, выше). Здесь мы также получили таблицу с
результатами работы BLAST (таблица 1, выше). Всего BLAST нашел 4 562 белка.
Некоторые результаты я отобрал для выравнивания. Всего для дальнейшего исследования я взял 17 белков, которые поделил на 3 группы:
-
1
выравнивание белка HCS_KITSK с наиболее схожими последовательностями. Процент соответствия (identity) у них выше 70%, E-value меньше 10-164. Также величина покрытия больше 95%. Здесь нет ни одного инделя (пропусков нет), у первых двух последовательностей даже нет несовпадений (возможно это те же самые белки). У остальных по два инделя. -
2
выравнивание белка HCS_KITSK с последовательностями средней степени схожести. Процент соответствия (identity) у них выше 30%, E-value меньше 10-41. Величина покрытия больше 94%. Гэпов достаточно много. -
3
выравнивание белка HCS_KITSK с последовательностями малой степени схожести. Процент соответствия (identity) у них выше 25%, E-value меньше 10-18. Величина покрытия больше 90%. Гэпов достаточно много.
Результат в таблице gryzunov_hits.xlsx
Выводы
Так как мой белок HCS_KITSK отвечает за вторичный метаболизм бактерий, то человеку известно не
так уж и много белков, сходных с моим. Вторичные метаболиты бывают очень разнообразными,
следовательно
и белки мало чем похожи друг на друга. Этот факт меня расстроил; также, он отразился на том, что
гомологичными я могу назвать только те белки, которые я использовал в первом выравнивании (рисунок 2,
выше).
Во втором (рисунок 3) и третьем (рисунок 4) выравниваниях получились очень
плохие результаты (в плане гомологии), хотя мы можем наблюдать интересные совпадения.
Например, колонки 50–65 во втором и 30–45 в третьем выравниваниях довольно похожи
W-[ER]-W-x(5)-L-x(5)-A-[RQ], колонки 103–107 во втором и 83–87 D-D-x(2)-D и так далее.
Белки из первого выравнивания (рисунок 2) я расцениваю как гомологичные на том основании, что мы видим множество консервативных колонок расположеных подряд. Например, с 52 по 66 колонку мы видим 15 полностью консервативных позиций. Также мы можем наблюдать полностью консервативные позиции с 76 колонки по 84, с 175 по 193, с 300 по 308 колонку и так далее.
Задание 2. Карта сходства двух белков
Что было сделано?
- Выбрали по идентификатору записи Uniprot из двух разных групп из данного заранее списка (всего групп 3);
- Построили карту локального сходства последовательностей этих белков;
- Объяснили крупные различия между последовательностями
Карта локального сходства
Для этого задания я выбрал два белка из разных груп с идентификаторами E4ZIZ7_LEPMJ и
K1VG04_TRIAC. У этих белков нет ручной аннотации на сайте Uniprot (для этих белков есть
только нерецензированные, автоматически аннотированные записи).
Затем получил последовательности этих белков в FASTA-формате и ввел их в текстовое поле Enter FASTA sequence(s), предварительно выставив флажок Align two or more sequences. Нажал на кнопку BLAST и запустил выравнивание двух белков. В результатах получил изображение выравнивания (рисунок 6) и карту локального сходства (Dot Matrix, рисунок 7).
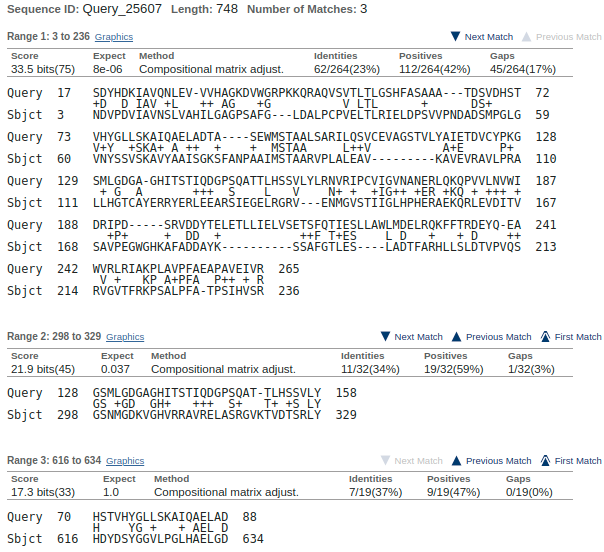
Рисунок 6.
Результат парного выравнивания двух аминокислотных последовательностей E4ZIZ7_LEPMJ и K1VG04_TRIAC в BLAST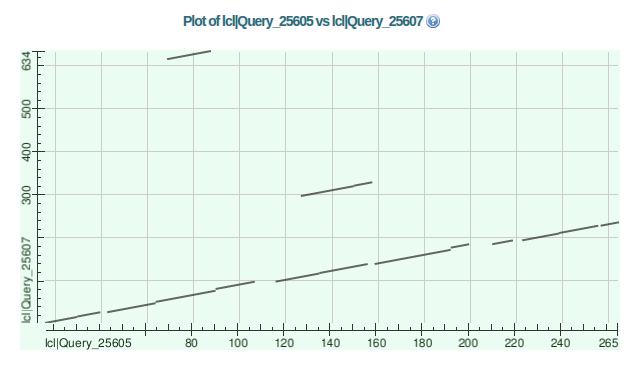
Рисунок 7.
Карта локального сходства для двух аминокислотных последовательностей E4ZIZ7_LEPMJ и K1VG04_TRIAC в BLAST. Query_25605 — E4ZIZ7_LEPMJ, Query_25607 — K1VG04_TRIAC.Выводы
Из карты локального сходства двух аминокислотных последовательностей E4ZIZ7_LEPMJ и K1VG04_TRIAС (рисунок 7) мы можем заключить, что разница между этими белками состоит в следующем. У последовательности K1VG04_TRIAС (Query_25607) относительно последовательности E4ZIZ7_LEPMJ (Query_25605) произошло две большие дупликации и три малые инсерции. У E4ZIZ7_LEPMJ относительно K1VG04_TRIAС произошло 6 инсерций.
Задание 3. Игры с BLAST
Что было сделано?
Сначала был произведен поиск случайной последовательности в BLAST. Для игры с параметрами BLAST мной была придумана последовательность Surely you're joking, Mr. Alexeevsky! . Затем я повторил первое задание, изменяя входные параметры BLAST.
Surely you're joking, Mr. Alexeevsky!
Эту последовательность я подал на вход программе blastp. На выходе были получены не самые лучшие результаты: количество очков — менее 40, процент покрытия — очень сильно варьируется от 90 до 30, E-value — от 3 до 265, соответствие (identity) — от 33 до 85. Все неизвестные символы BLAST заменял на X.
Изменение входных параметров BLAST
На этот раз я использовал последовательность белка HCS_KITSK. Я производил поиск несколько раз,
каждый раз изменяя какой-нибудь один параметр.
Сначала я использовал стандартные параметры и получил 3346 результатов. Затем я попробовал
отключить параметр Short queries, тогда я получил 3562 . Список топ–100 результатов
не изменился, как и его "свойства" (E-values и так далее). Затем я поставил параметр
Expect threshold на 0,001. И получил 3033 результатов, в то же время свойства топ–100 результатов
не изменились.
После я изменил Word size на 2. На выходе я получил 7187 результатов. Опять же,
свойства топ–100 результатов не изменились.
Далее выбрал Matrix –
BLOSUM90, на выходе получил 3339 результатов. Это изменение довольно сильно повлияло на
E-values не начальных последовательностей, что говорит, что похожесть этих последовательностей
очень зависит от ситуации, в которой они были оценены. Когда же переставил этот параметр на
BLOSUM45, то получил 5028 результатов. Сразу стало заметно, что E-values стали меньше на 10
порядков. Также я решил испытать матрицы серии PAM–(point accepted mutation matrix).
Попробовал для начала PAM30, получил достаточно малое количество результатов – всего 890.
E-values увеличились на 20 порядков. На процент идентичности это изменение влияния не
оказало. Я также решил испробовать PAM250. Получил 3537 результатов, причем происходит
смена мест в топ-10 результатов, E-values увеличиваются в 20 раз, процент соответствия (identity)
незначительно падает (на 1–2%)
Напоследок я решил испробовать поля Low complexity regions и Compositional adjustments. Когда я убрал флажок с первого поля, то я получил 3528 результатов, одновременно с этим E-values незначительно выросли. Когда я испытывал последний параметр (выставив No adjustment), то я получил 3064 результатов, опять же, E-value которых было несколько больше.