Семестр 7, автор – Грызунов Никита (nikit00000s)
Практикум 4. Валидация.
Задание 1.
Мне была выдана запись PDB с идентификатором 1SBP. Она описывает белок, специфически связывающий сульфат и участвующий в трансмембранном транспорте сульфата у грамотрицательной бактерии Salmonella typhimurium. Он локализован в периплазматическом пространстве.
Основные параметры записи 1SBP:
- эксперимент: РСА;
- организм: Salmonella enterica subsp. enterica serovar Typhimurium;
- год: 1993;
- разрешение: 1.70 Å;
- R-value observed: 0.178;
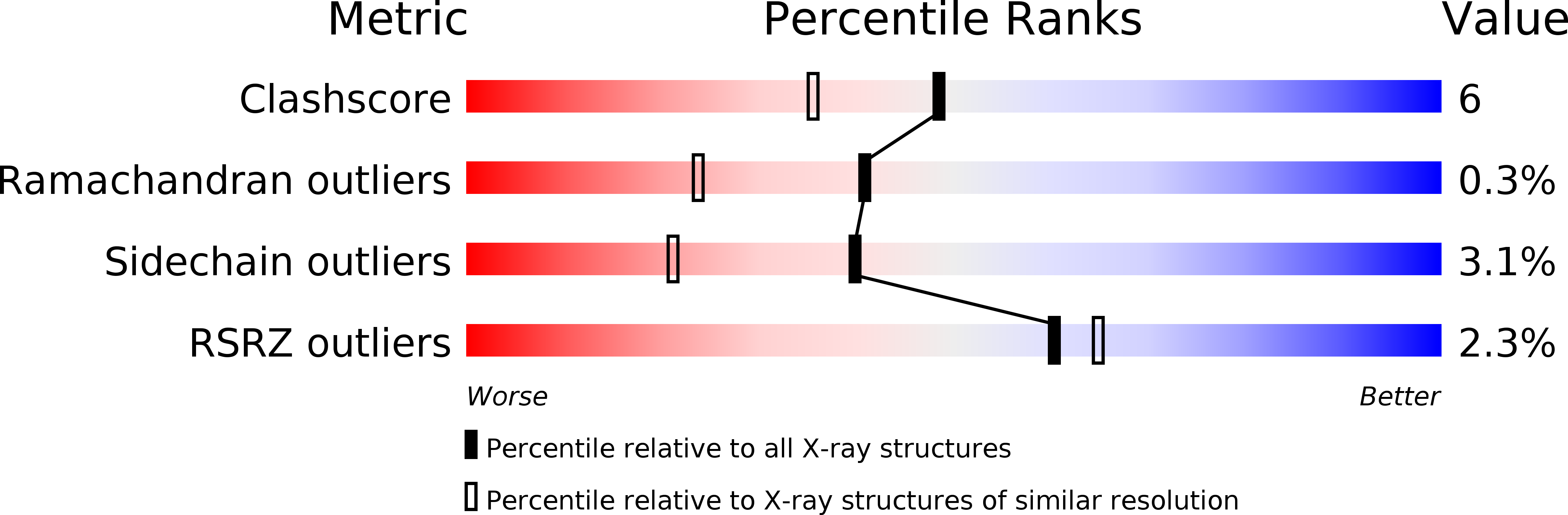
- clashscore: 6;
- Ramachandran outliers: 0.3%;
- side-chain o.: 3.1%;
- RSRZ o.: 2.3%.
Также приведем информативную картинку wwPDB Validation (рис. 1):
Приведем интересующие нас поля "REMARK":
REMARK 3 DATA USED IN REFINEMENT. REMARK 3 RESOLUTION RANGE HIGH (ANGSTROMS) : 1.70 REMARK 3 RESOLUTION RANGE LOW (ANGSTROMS) : NULL REMARK 3 DATA CUTOFF (SIGMA(F)) : NULL REMARK 3 COMPLETENESS FOR RANGE (%) : NULL REMARK 3 NUMBER OF REFLECTIONS : NULL
REMARK 3 FIT TO DATA USED IN REFINEMENT. REMARK 3 CROSS-VALIDATION METHOD : NULL REMARK 3 FREE R VALUE TEST SET SELECTION : NULL REMARK 3 R VALUE (WORKING + TEST SET) : 0.178 REMARK 3 R VALUE (WORKING SET) : NULL REMARK 3 FREE R VALUE : NULL REMARK 3 FREE R VALUE TEST SET SIZE (%) : NULL REMARK 3 FREE R VALUE TEST SET COUNT : NULL REMARK 3 REMARK 3 FIT/AGREEMENT OF MODEL WITH ALL DATA. REMARK 3 R VALUE (WORKING + TEST SET, NO CUTOFF) : NULL REMARK 3 R VALUE (WORKING SET, NO CUTOFF) : NULL REMARK 3 FREE R VALUE (NO CUTOFF) : NULL REMARK 3 FREE R VALUE TEST SET SIZE (%, NO CUTOFF) : NULL REMARK 3 FREE R VALUE TEST SET COUNT (NO CUTOFF) : NULL REMARK 3 TOTAL NUMBER OF REFLECTIONS (NO CUTOFF) : NULL
Как можно видеть, в процессе оптимизации структуры не использовали R-free.
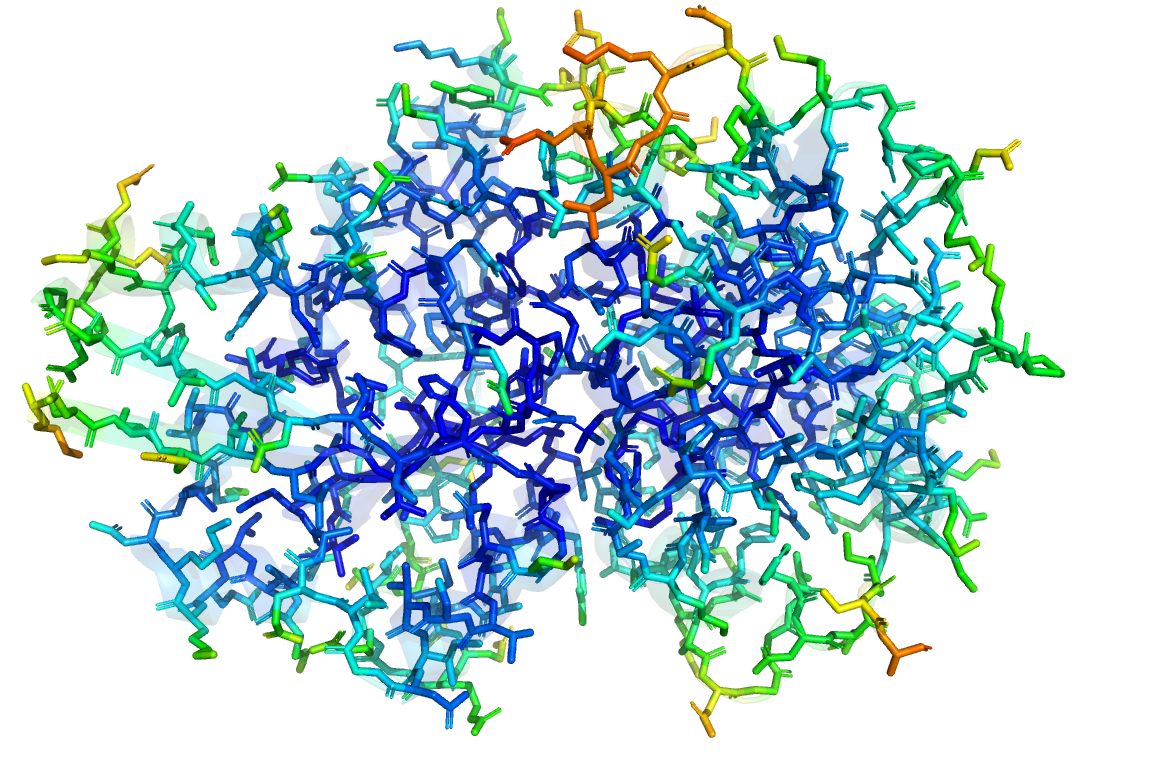
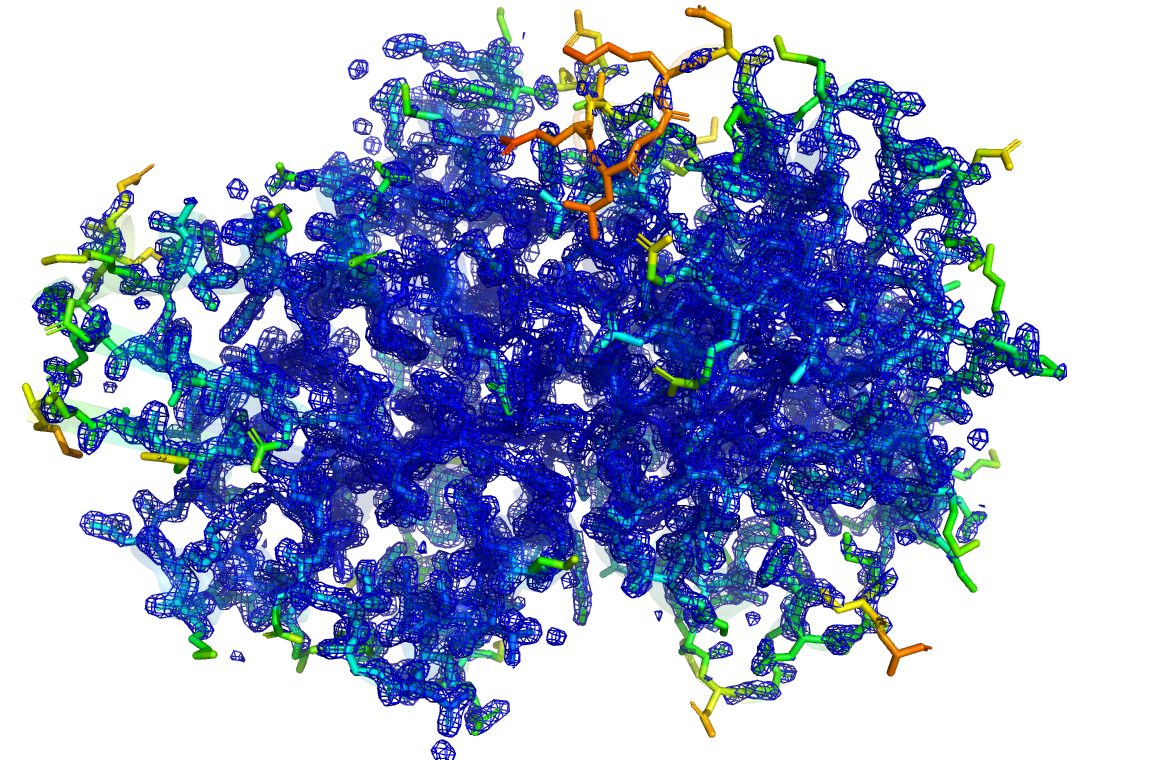
Как можно заметить из рис. 2, аминокислоты, не имеющие поддержки ЭП, в основном находятся на поверхности структуры и обладают меньшими значениями B-фактора (рис. 3). Естественно, что большинство из таких остатков являются полярными (и даже заряженными). Внутри глобулы аминокислот без поддержки ЭП обнаружено не было.
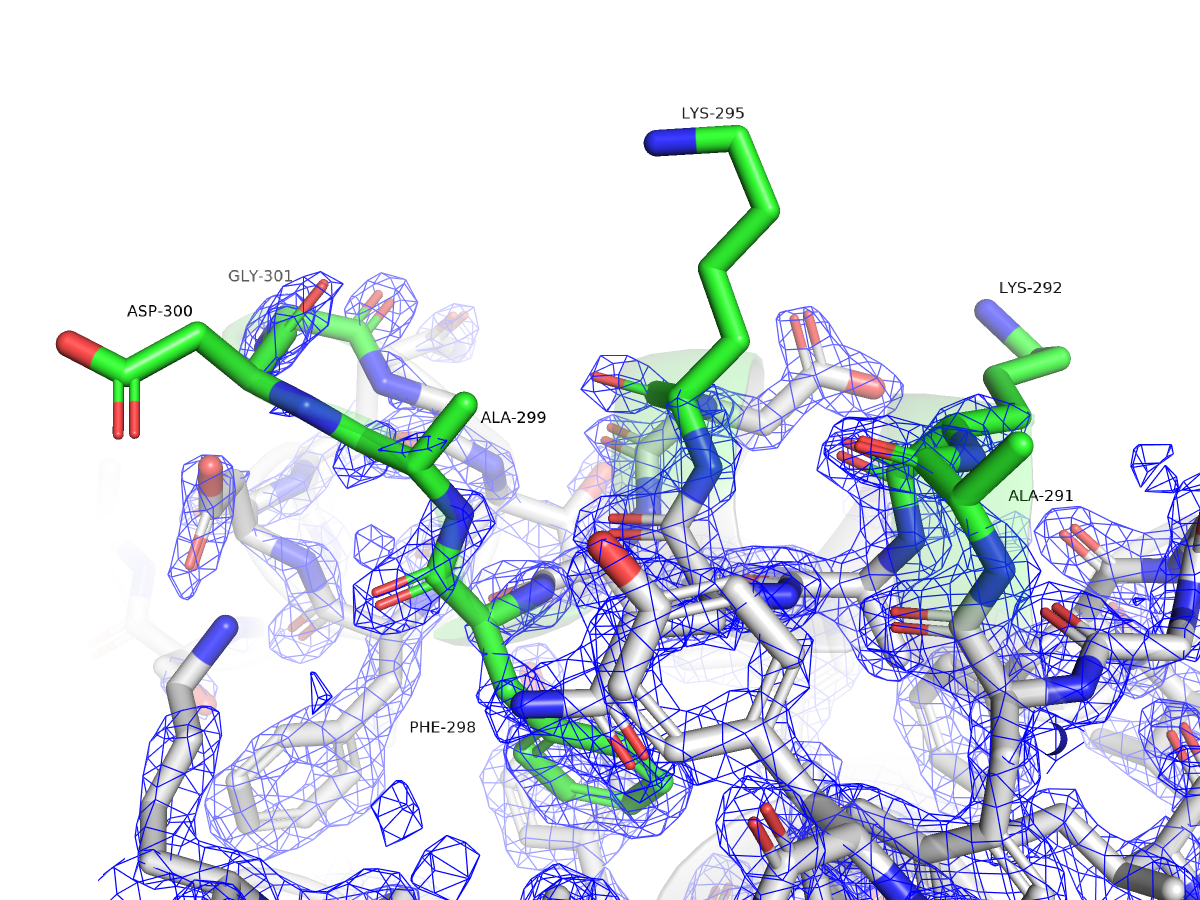
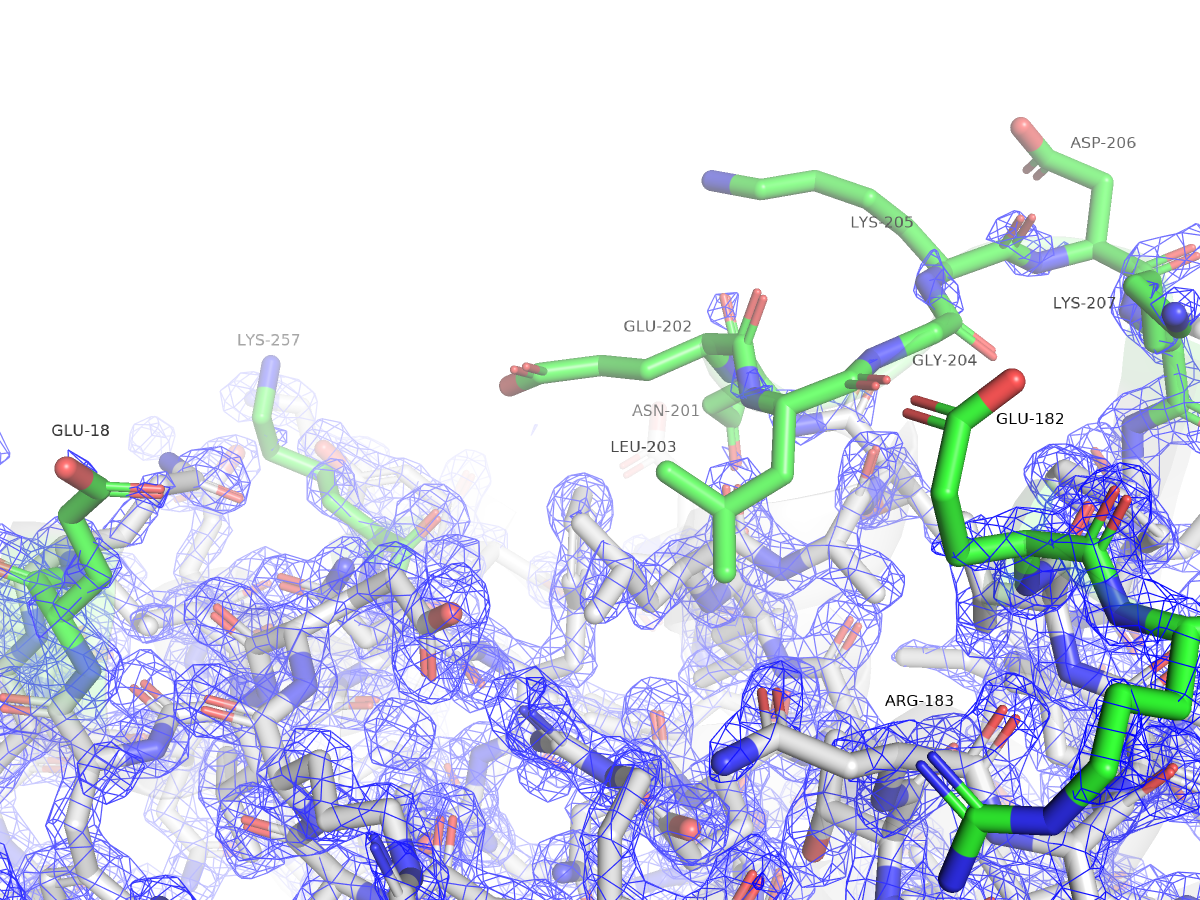
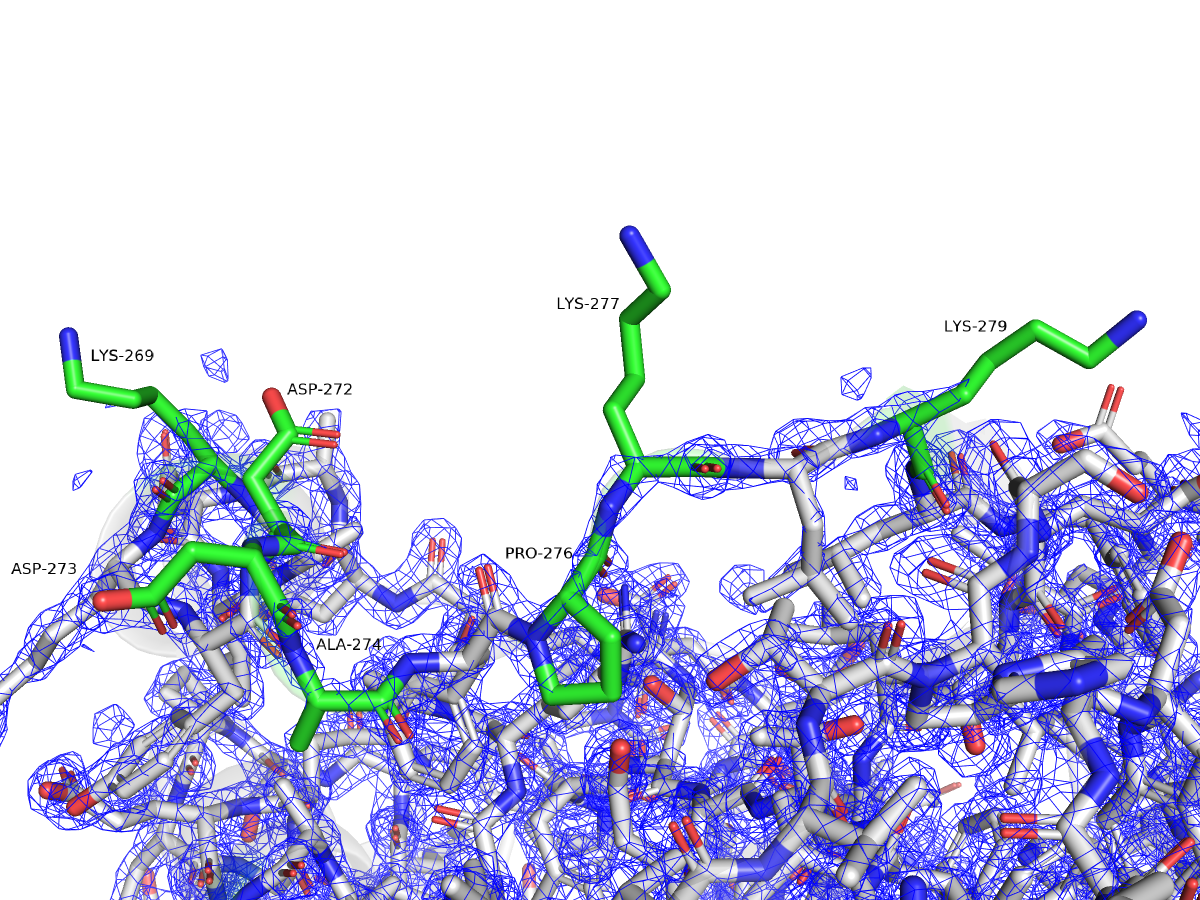
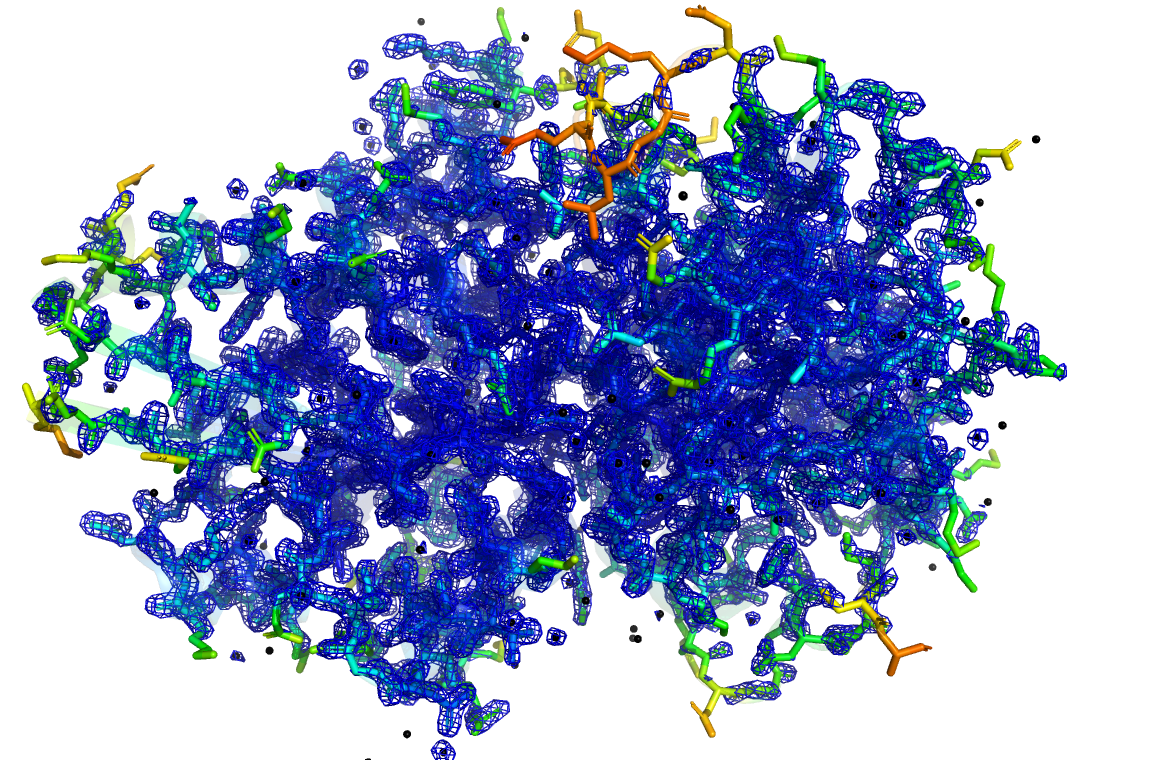
Проверим, есть ли альтернативные структуры для данного пептида (рис. 4).
В целом, разрешение структуры под идентификатором 1SBP не такое уж и плохое, и составляет 1.70 Å. Значения метрик wwPDB Validation - средние. Альтернативных экспериментальных структур, к сожалению, не существует, только структура, предсказанная AlphaFold, поэтому предпочтительно использовать именно данную структуру 1SBP.
Задание 2.
Воспользуемся специальными инструментами и вычислениями, чтобы валидация структуры не ограничилась оценкой "на глаз". Для поиска маргинальных остатков можно использовать обширный инструмент Molprobity, однако, к моменту выполнения практикума он не работает, поэтому будут использованы несколько других инструментов, способных воспроизвести функционал оригинального инструмента. Первый инструмент - SwissModel Structure Assessment. Результат оценки структуры с помощью SwissModel состоит из многих компонент. Во-первых, это оценка торсионных углов главной цепи протеина.
SwissModel Structure Assessment
Карты Рамачандрана
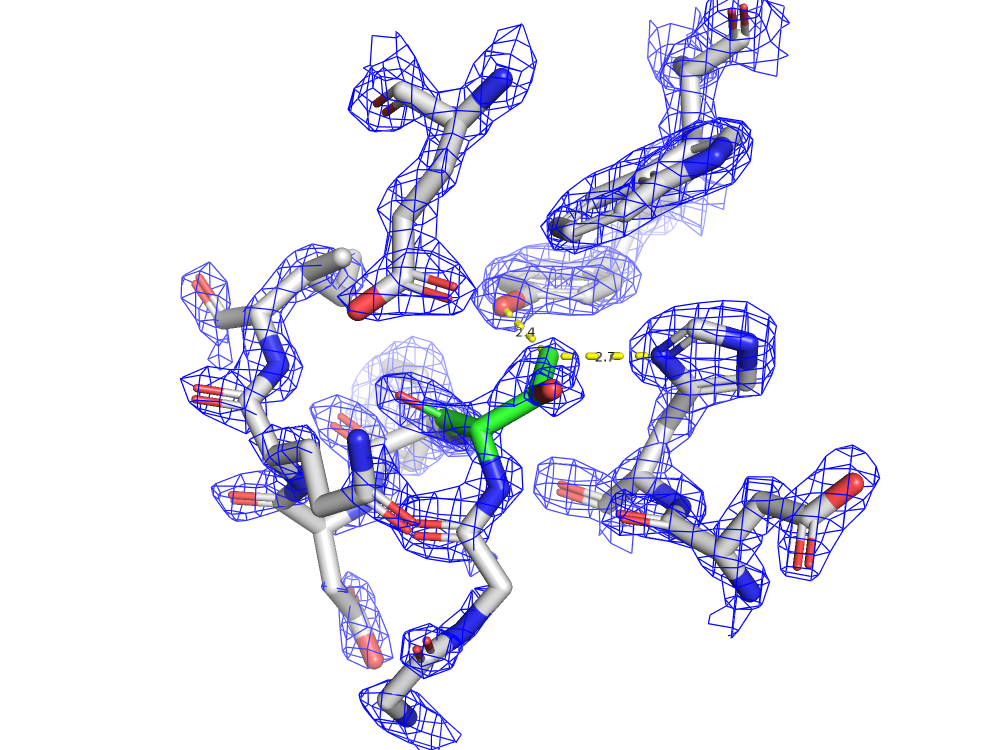
Приведем небольшую литературную справку по тому, почему стоит различать остатки при рассмотрении карт Рамачандрана. Наибольшее количество разрешенных конформаций (углов φ и ψ) наблюдается у остатков глицина (рис. 6, B). Главная запрещенная область - вертикальная, сохраняющаяся вдоль всех значений ψ при значениях φ, близких к нулю, вызвана отталкиванием С'i-1 и С'i атомов, так как минимальное расстояние между ними - rmin= 3,0 Å для C...C взаимодействия. Схожий стерический запрет есть и в горизонтальной области (ψ, близкие к нулю, многие φ), но он уже вызван отталкиванием Ni-1 и Ni атомов - rmin= 2,7 Å для N...N взаимодействия, что меньше, чем в предыдущем случае [1].
Из этого всего можно заключить, что по углу φ вращаться труднее, чем по углу ψ. Но если бы дело ограничивалось только этими взаимодействиями, вращения по двум углам были бы не взаимосвязанны, и запретные области были бы ограничены только вертикальной и горизонтальной полосами вдоль осей координат (рис. 5, A). Ситуация меняется, если учесть, что при атоме C′ есть еще О- и Cα-атомы, а при атоме N есть также H- и C′-атомы. Тогда получится карта самого маленького аминокислотного остатка - глицина (боковой радикал -H, рис. 5. B)
У всех остальных аминокислотных остатков радикал куда больше, и столкновение их Cβ-атомов с C′i–1 сильно ограничивает разрешенную область по углам φ, а с Ni+1 - по углам ψ. Так как сталкивается именно Cβ атом, то ситуация у большинства остатков схожая (рис. 5, C).
Интересна ситуация с иминокислотой пролин. У него угол φ практически фиксирован кольцом при -70°, а вращение по углу ψ — такое же, как у аланина (рис. 5, D). Кроме того, кольцо пролина сужает область разрешенных конформаций остатка, лежащего перед ним в цепи (рис. 5, E).
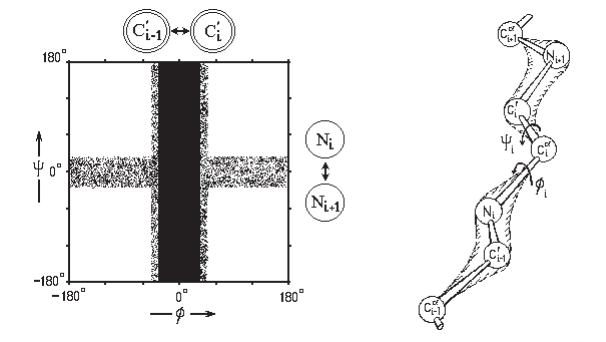
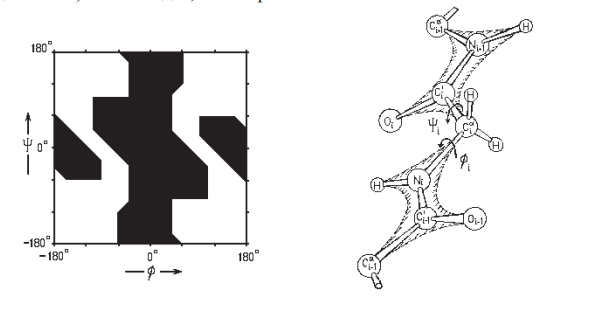
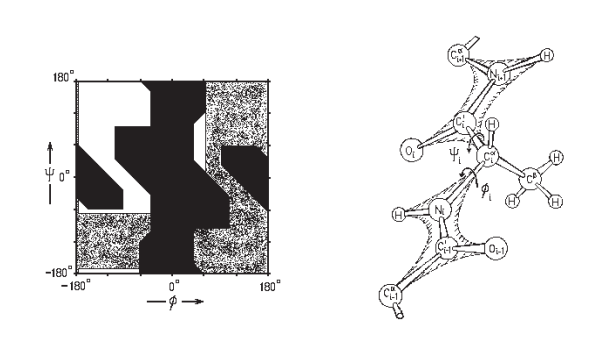
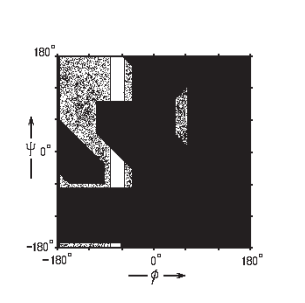
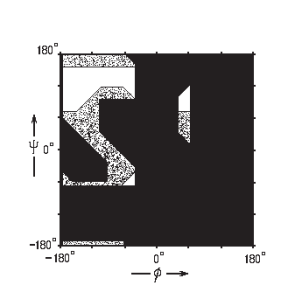
Вернемся к картам Рамачандрана, полученными с помощью SwissModel для структуры 1SBP. Они разбиты на несколько групп аминокислот из-за их особенностей, что объяснялось ранее (рис. 6).
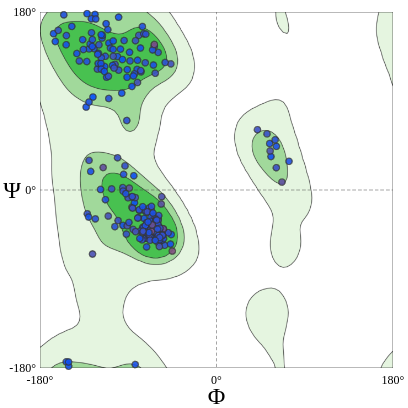
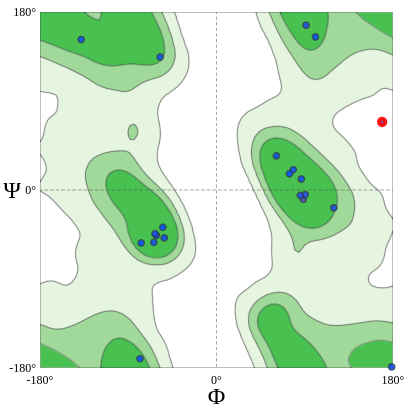
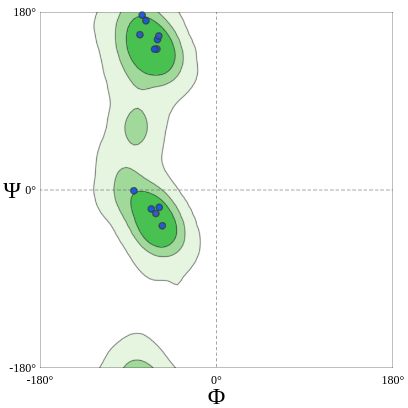
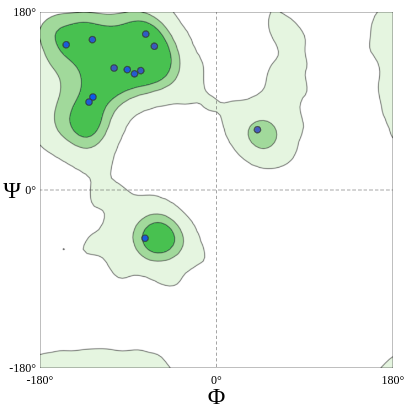
Если ссылаться на литературные источники, то можно заключить, что практически все остатки структуры 1SBP находятся в разрешенной области карт Рамачандрана. Кроме того, если сослаться на документацию SwissModel Structure Assessment, то можно заключить, что качество структуры 1SBP также подтверждено тем, что значения углов остатков этой структуры находятся в областях карт с высокой плотностью (там наблюдается высокое количество остатков из выборки 12,521 экспериментальных структур, то есть эти карты поддерживаются большим количеством экспериментальных структур).
Всего 98.37% всех остатков имеют удовлетворительную конформацию главной цепи, и только один остаток является маргинальным с точки зрения карт Рамачандрана - остаток GLY`204 (рис. 7).
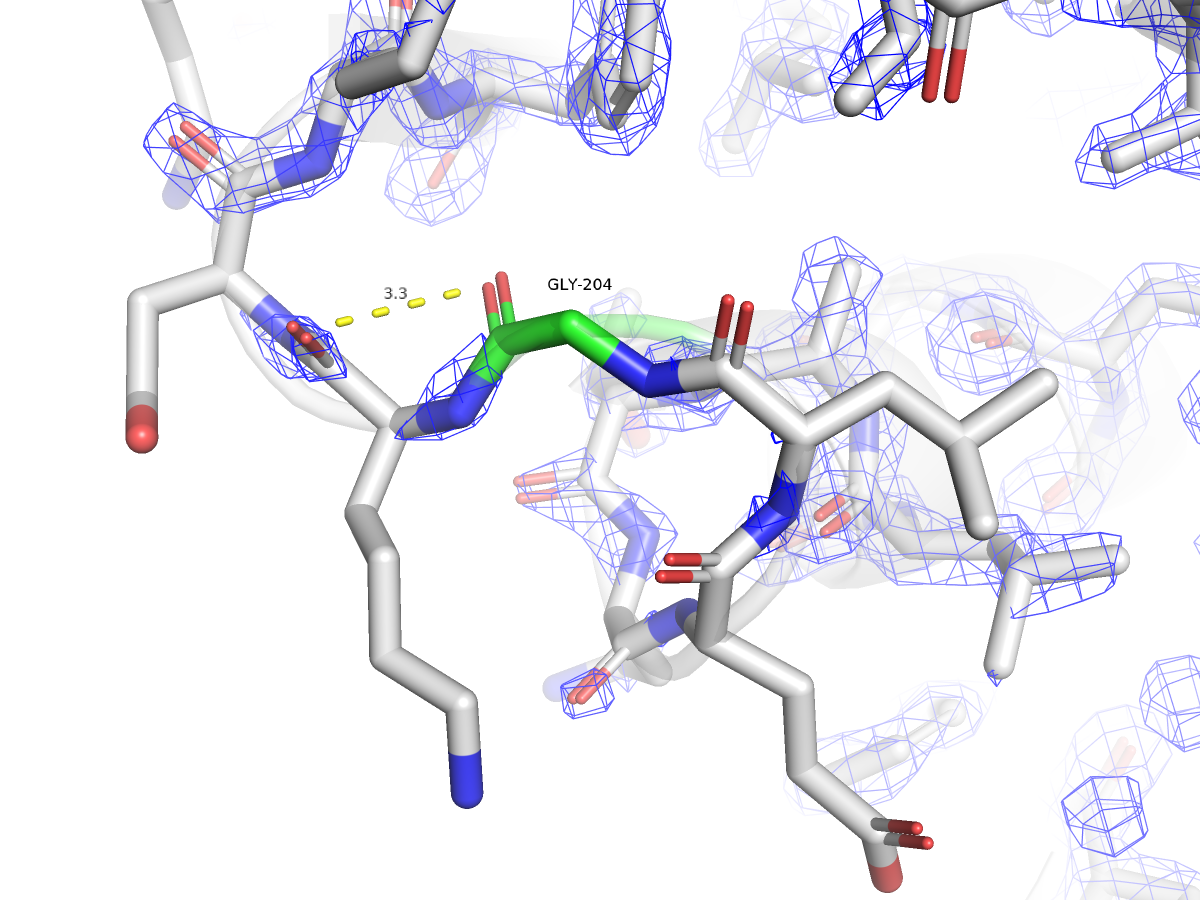
Результаты MolProbity
Приведем результаты в таблице 1 (пояснения приведены из документации SwissModel Structure Assessment):
| Свойство (метрика) | Значение | Остатки | Пояснение |
|---|---|---|---|
| MolProbity Score | 1.87 | Совокупное качество структуры, отражающее при каком разрешении эксперимента РСА ожидалась бы структура такого же качества (в нашем случае, качество структуры ниже ожидаемого для разрешения 1.7 Å) | |
| Clash Score | 6.04 | (A137 TYR-A303 THR), (A297 HIS-A303 THR), (A19 GLN-A258 ASN) | Под перекрытиями (clashes) подразумевается сближение отдельных атомов на 0,45 Å без образования водородной связи |
| Ramachandran Favoured | 98.37% | Процент остатков, удовлетворяющих разрешенным конформациям главной цепи | |
| Ramachandran Outliers | 0.33% | A204 GLY | Процент остатков, неудовлетворяющих разрешенным конформациям главной цепи. При разрешениях ниже 3,0 Å любые выбросы следует считать ошибками. |
| Rotamer Outliers | 5.10% | A65 LEU, A206 ASP, A203 LEU, A74 GLU, A164 LYS, A303 THR, A60 THR, A300 ASP, A32 THR, A4 GLN, A80 LYS, A205 LYS, A269 LYS | Процент остатков, неудовлетворяющих разрешенным конформациям боковых цепей. При разрешениях ниже 3,0 Å любые выбросы следует считать ошибками. |
| C-Beta Deviations | 10 | Положение отклоняется от идеального более чем на 0,25 Å (дословный перевод документации, больше объяснений далее в тексте). | |
| Bad Bonds | 4 / 2486 | A271 TYR-A272 ASP, A44 GLY, A172 GLY, A271 TYR | > 4σ отклонений от идеального значения |
| Bad Angles | 61 / 3376 | A32 THR, A60 THR, A303 THR, (A271 TYR-A272 ASP), A39 ASP, A110 HIS, A2 ASP, A183 ARG, A113 ASN, A19 GLN, A4 GLN, A27 HIS, (A75 ARG-A76 GLY), A206 ASP, A152 GLN, A102 ARG, A42 HIS, A227 ASP, (A141 TRP-A142 GLY), A169 LEU, A54 ASN, A79 ASP, A240 GLU, A149 ASN, A191 ALA, A14 ARG, A163 PHE, A96 THR, A10 TYR, A250 GLU, A182 GLU, A231 GLU, A84 LYS, (A164 LYS-A165 ASN), A74 GLU, A281 PHE, A15 GLU, (A207 LYS-A208 PHE), A171 SER, A190 ILE, (A107 LYS-A108 GLN), A235 THR, A67 TYR, (A223 VAL-A224 SER), A50 THR, A174 ARG, (A246 LEU-A247 TYR), (A171 SER-A172 GLY), A247 TYR, A234 ASP, A265 ALA | > 4σ отклонений от идеального значения |
Отклонение Cβ атома чувствительно к несоответствиям между боковыми и главными цепями, вызванными плохо подобранными (расшифрованными) конформациями или неподходящими параметрами улучшения модели. Так как длины связей сильно ограничены в разбросе значений, отклонения вокруг Cα вызваны отклонениями в углах связей. Угол N-Cα-C', в основном, определяется главной цепью, в то время как углы N-Cα-Cβ и C'-Cα-Cβ определяются совместимостью боковых и главной цепей. Информацию об этих двух углах можно "сжать" до простой метрики отклонения Cβ атома от его идеальной позиции, которая бы удовлетворяла углам N-Cα-Cβ и C'-Cα-Cβ и длине связи Cα-Cβ [2].
Возвращаясь к выводам SwissModel, можно заметить, что один остаток появляется во многих категориях - THR`303 (Clash Score, Rotamer Outliers, Bad Angles).
Quality Estimate
Последний вывод, полученный с помощью SwissModel, который мне хотелось бы привести - график Quality Estimate.
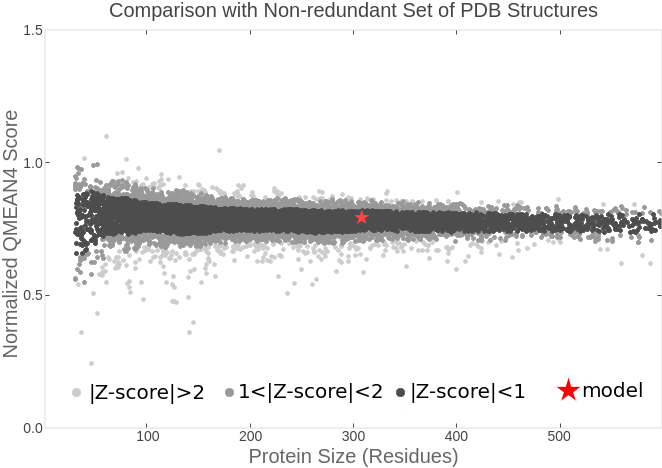
QMEAN4 - оценочная функция, являющаяся линейной комбинацией стандартизированных оценок (Z-score) 4 knowledge-based potentials (statistical potential) - псевдопотенциалов, основанных на статистическом анализе отдельных геометрических показателей (как локальных, так и глобальных) структур всего банка (источник).
QMEAN4 отдельной модели сравнивается с тем, что можно было бы ожидать от экспериментальных структур аналогичного размера. Она показывает, на сколько стандартных отклонений от среднего для экспериментальных структур данного размера отличается оценка данной модели.
Структура 1SBP, согласно QMEAN4, является довольно достоверной экспериментальной структурой, ведь она показывает примерно такое же качество, как и остальные экспериментальные структуры такого же размера (эта структура заслуживает доверия).
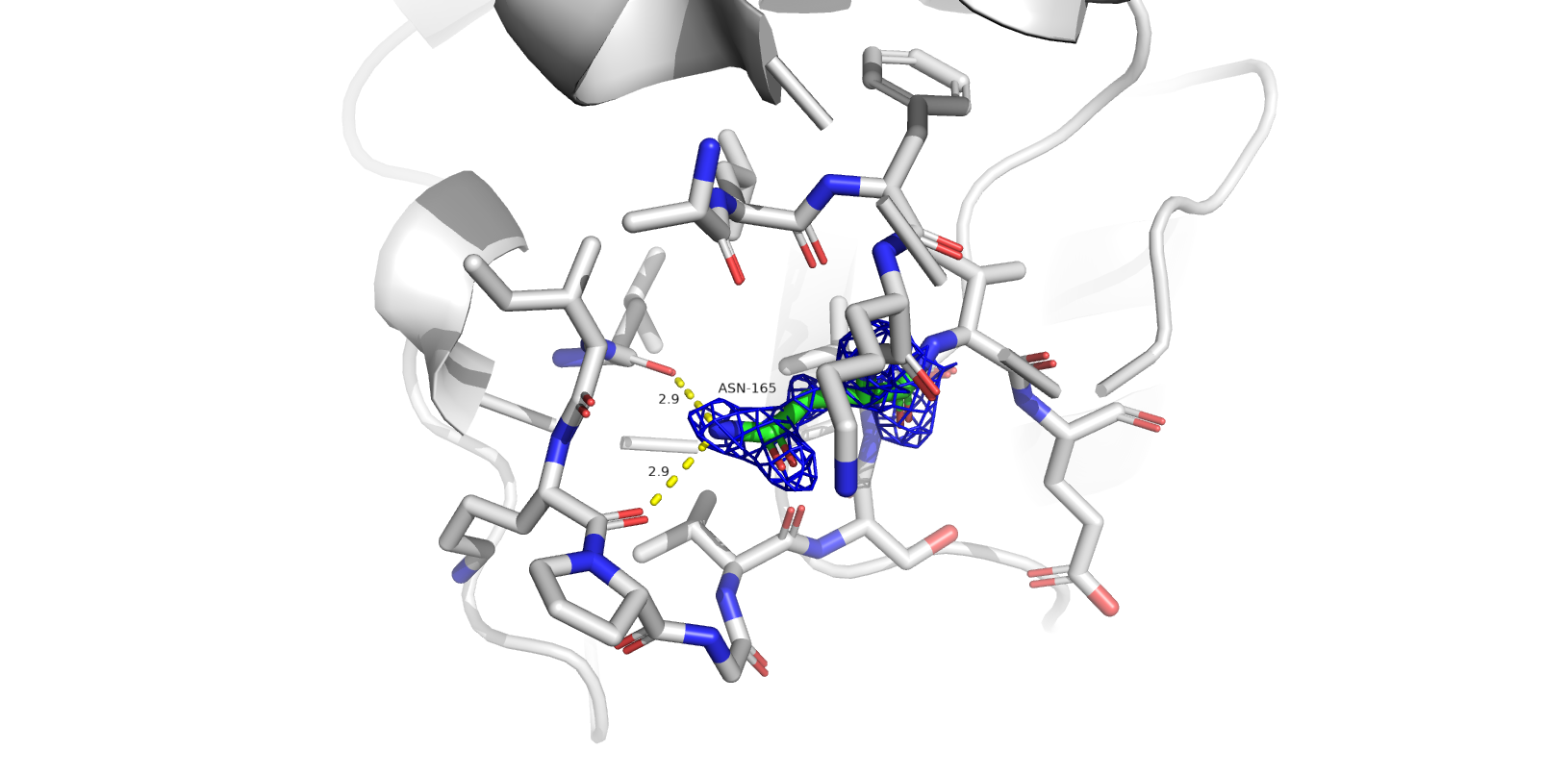
NQ-Flipper
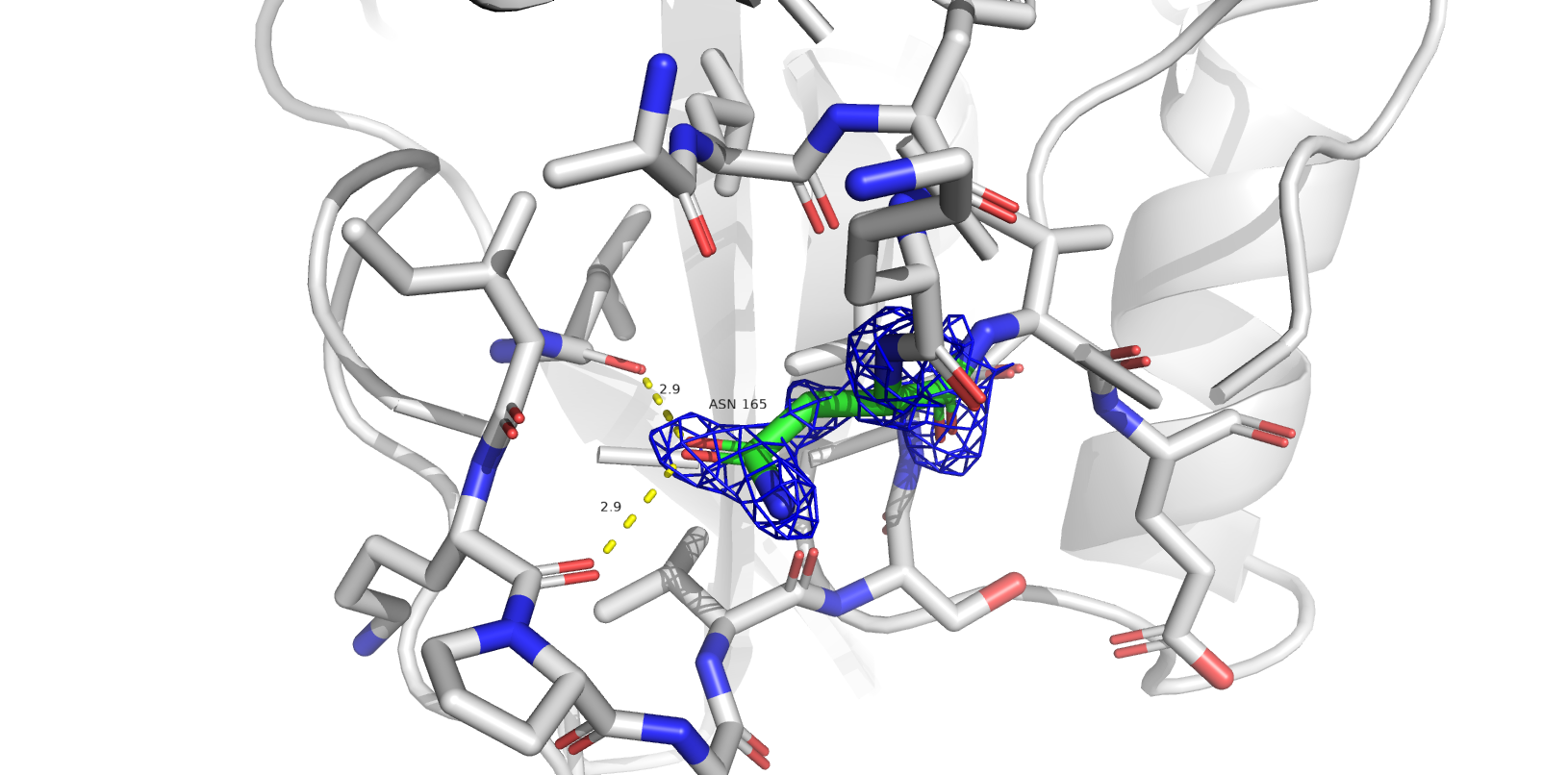
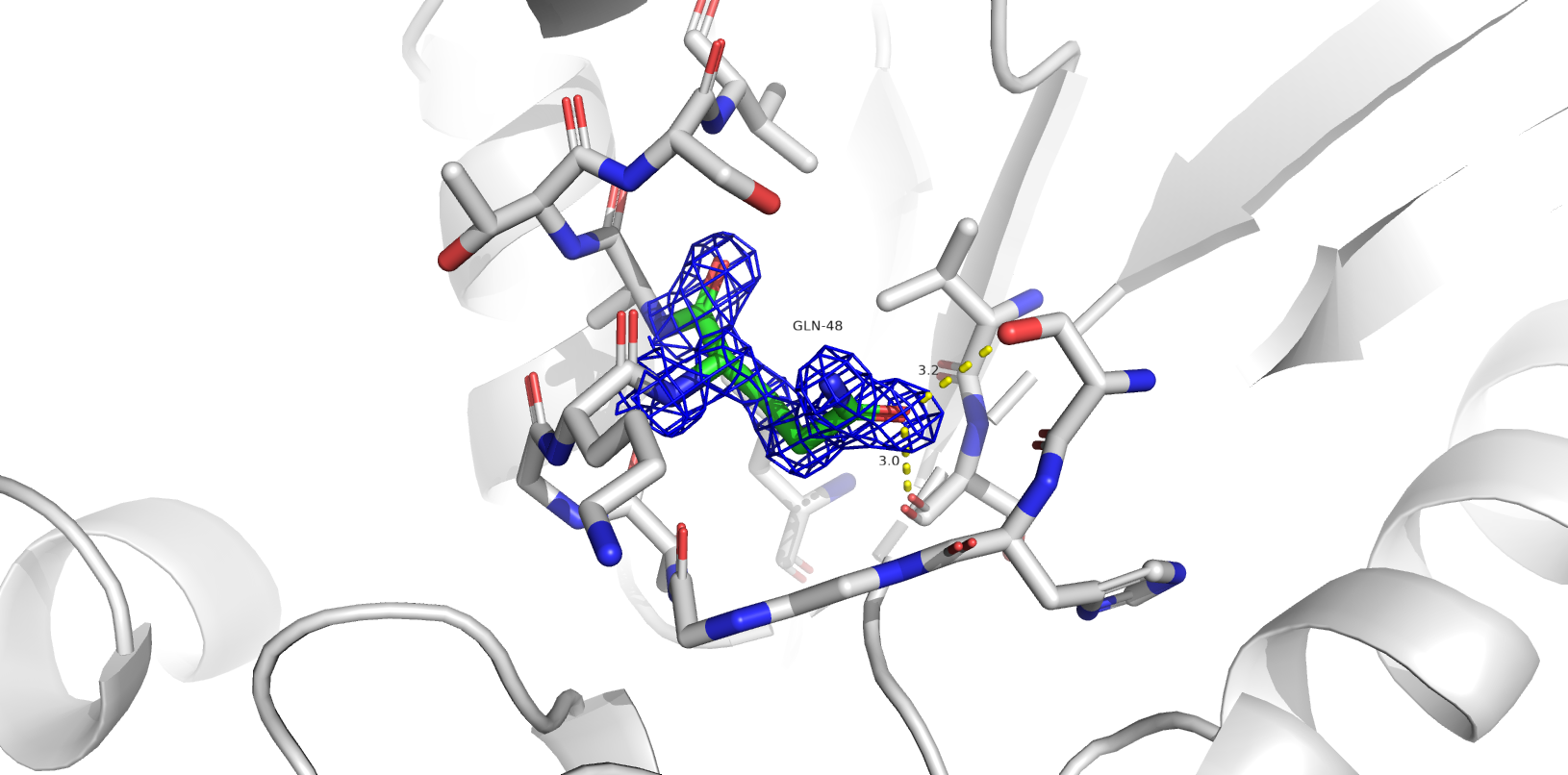
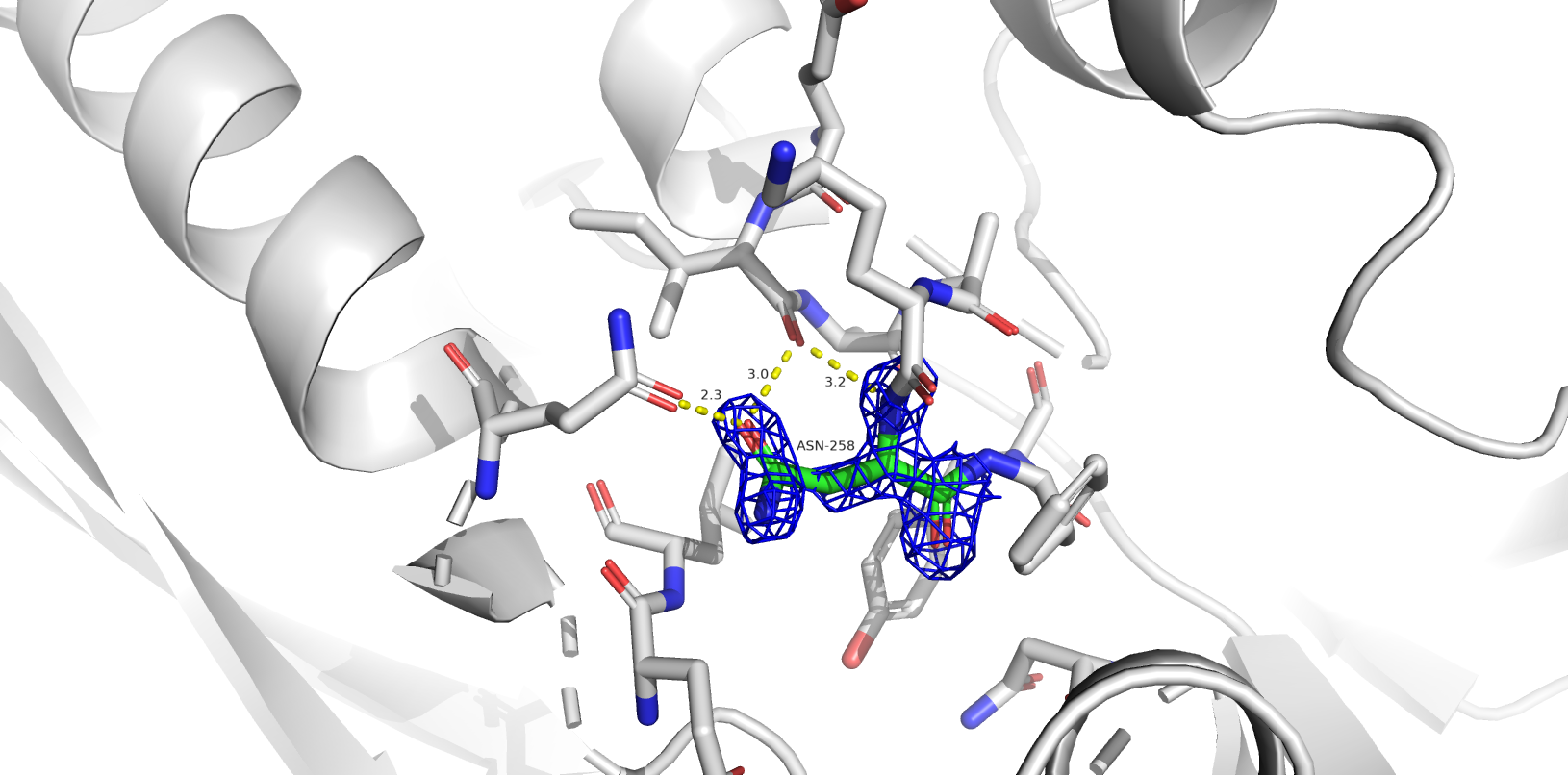
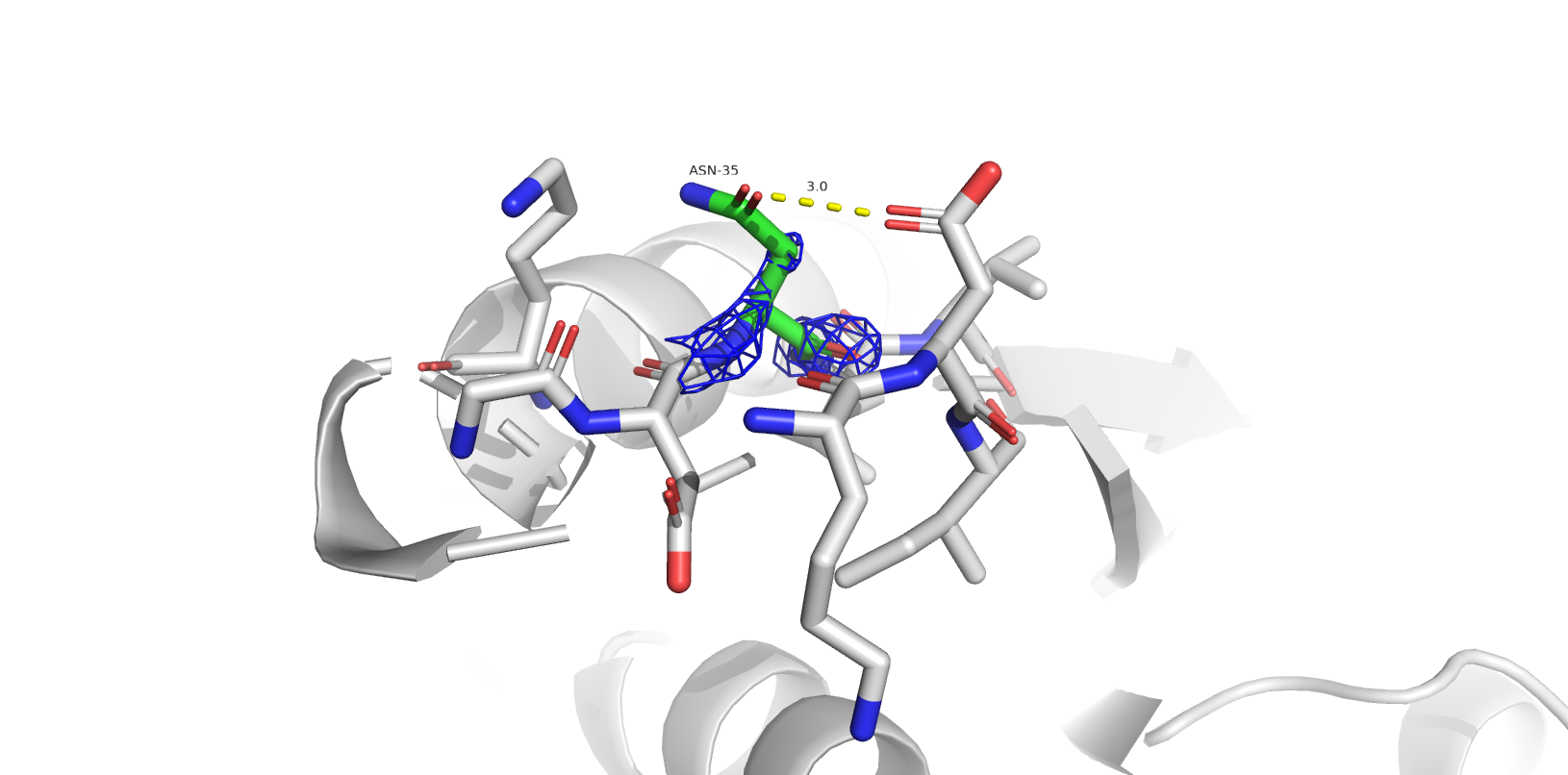
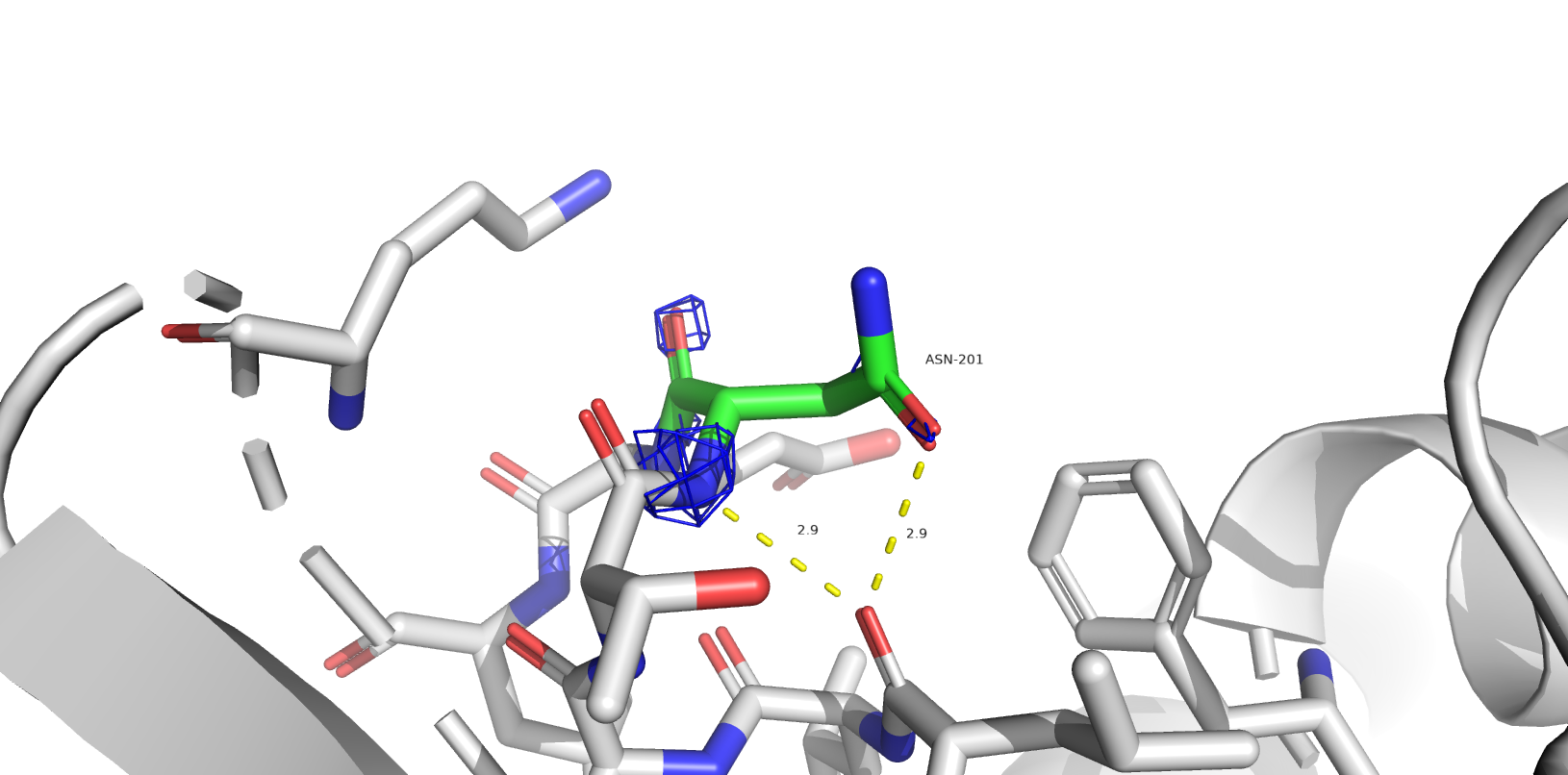
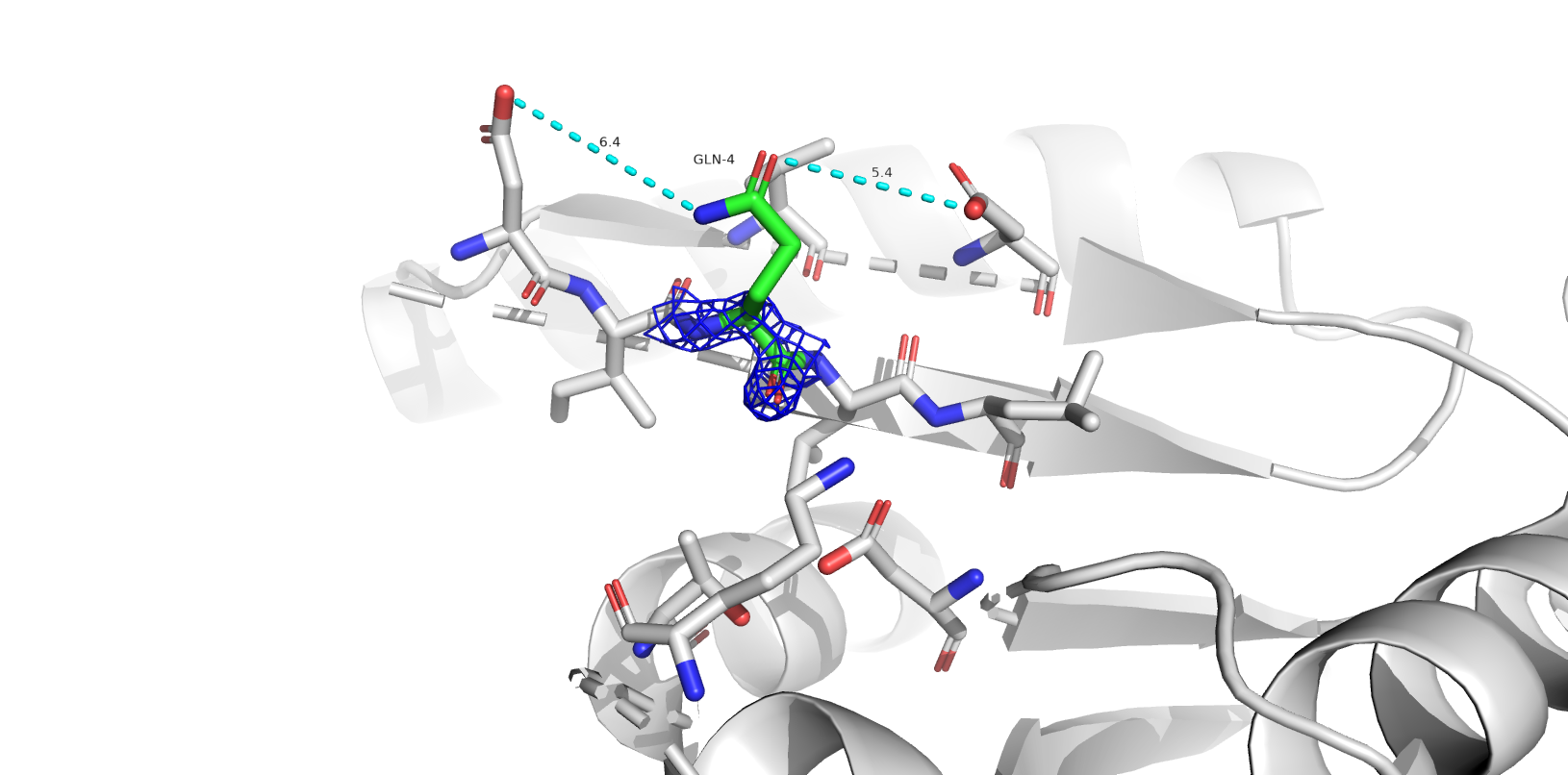
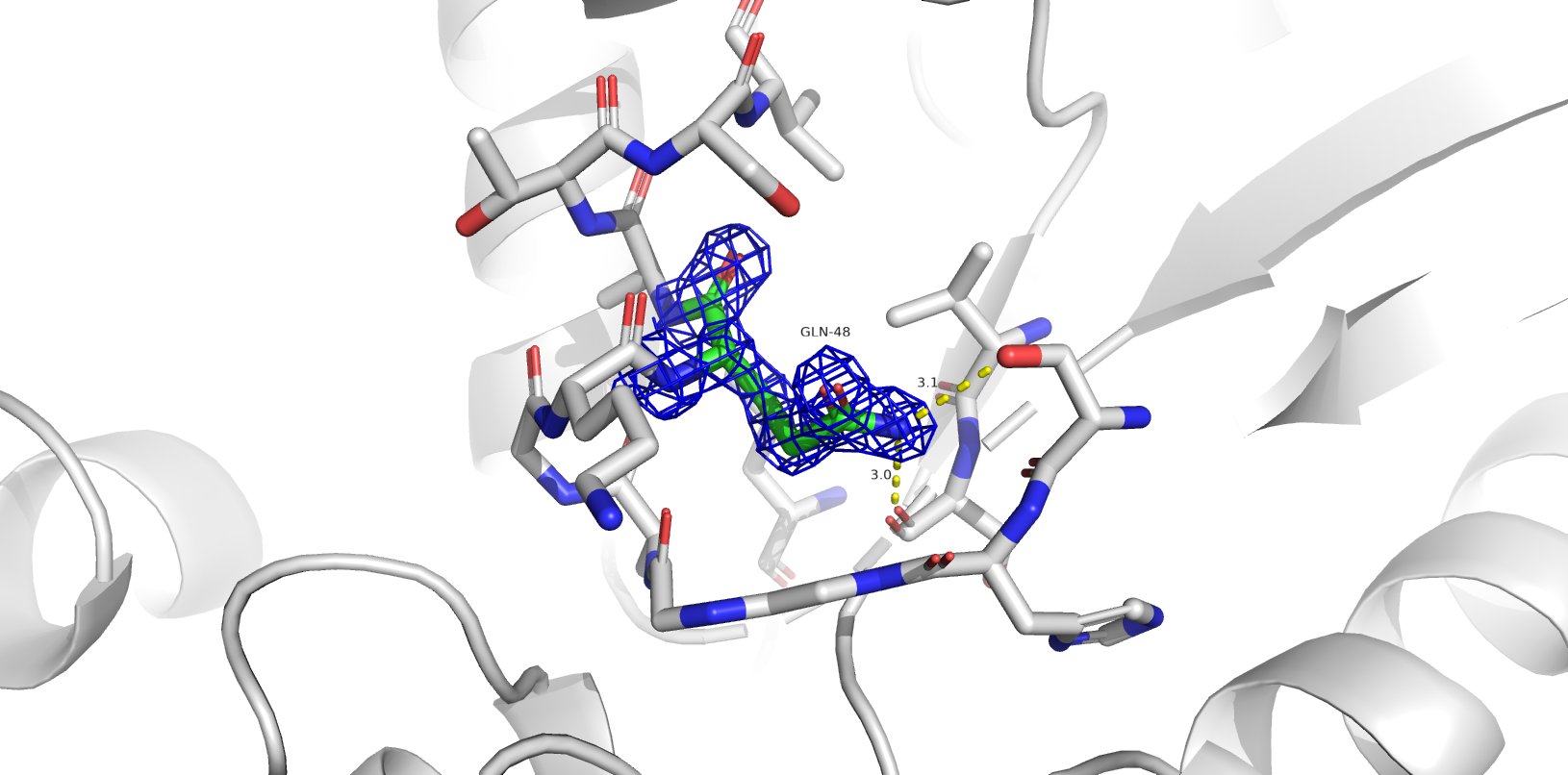
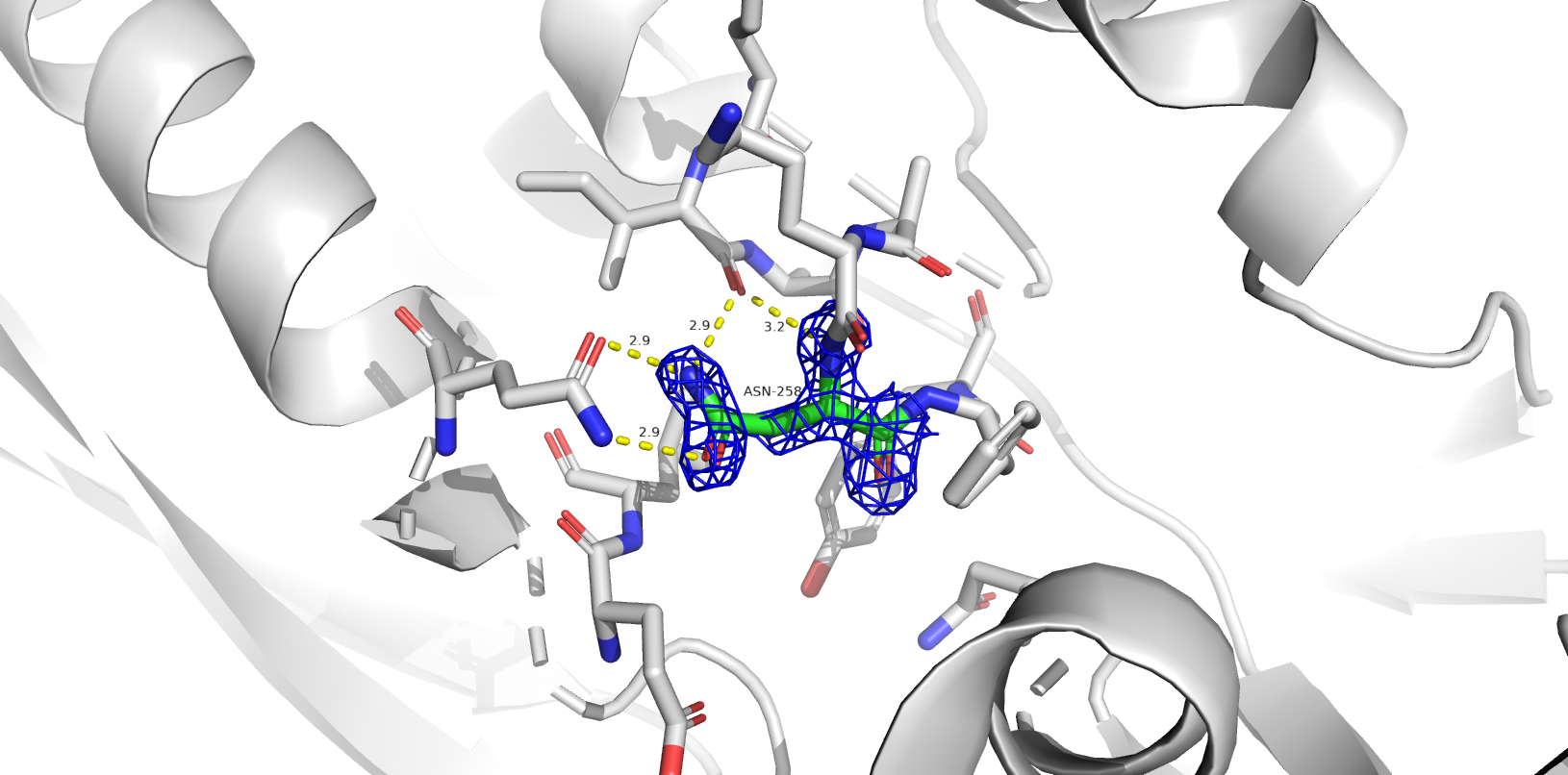
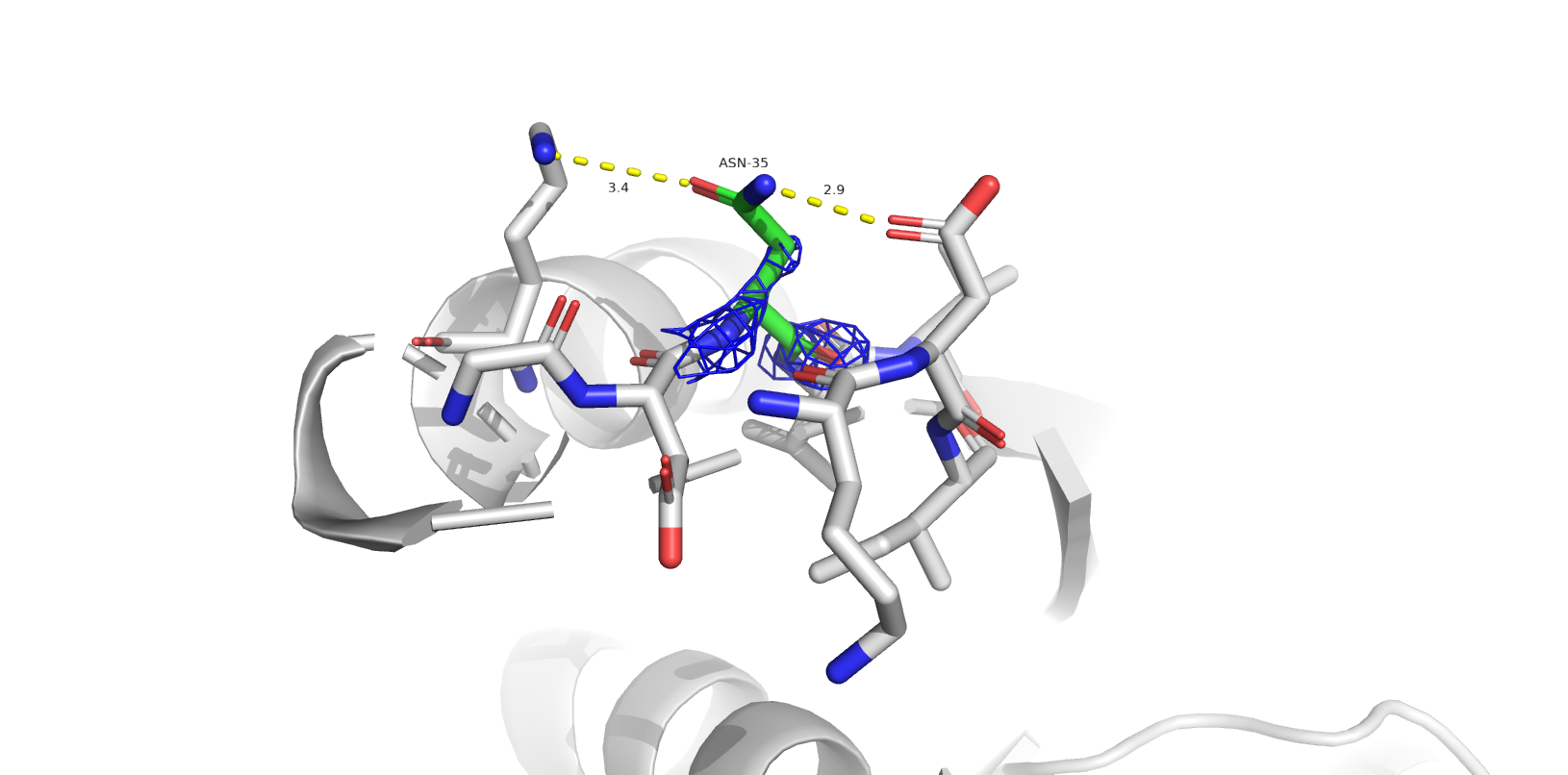
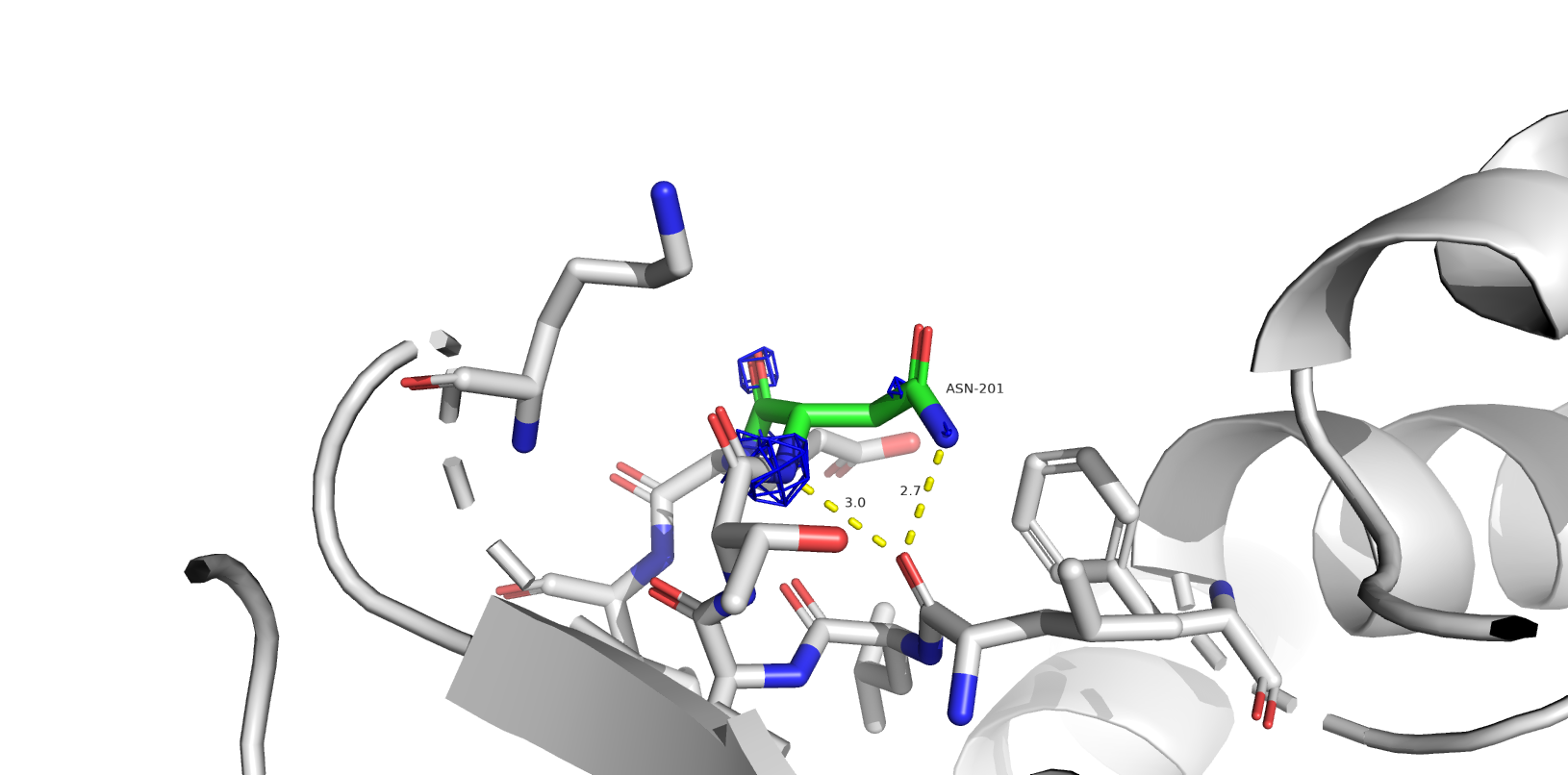
Одной из частых проблем программ, составляющих модель по электронной плотности, является переворачивание амидных групп боковых цепей глутамина и аспарагина. В структуре 1SBP программа нашла 6 значимых остатков: ASN`165, GLN`48, ASN`258, ASN`35, ASN`201, GLN`4 (в порядке значимости).
Ионов металлов в структуре не нашлось, поэтому инструмент Checkmymetal не пригодился.
В целом, можно сказать, что качество структуры - довольно хорошее, однако она требует некоторых исправлений.
- GLY`204 (torsion) - возможно, проблема заключается в том, что из-за того, что этот остаток находится на поверхности белка, в петле, то он более сильно подвержен тепловому движению, и из-за этого происходит неточность расшифровки РСА.
- THR`303 (clashes, bad angles) - возможно, здесь необходим разворот связи на 180° между Cα и Cβ (тогда -OH и -CH3 группы тирозина поменяются местами). После этого, вероятно будет возможно образование дополнительных водородных связей.
- ASN`165, GLN`48, ASN`258, ASN`35, ASN`201 (flip) - скорее всего, остатки некорректно ориентированы.
- GLN`4 (flip?) - скорее всего, от той или ориентации остатка ничего не поменяется.
Скорее всего, все эти неточности связаны с ошибками расшифровки РСА, а не с особенностями структуры.
Задание 3.
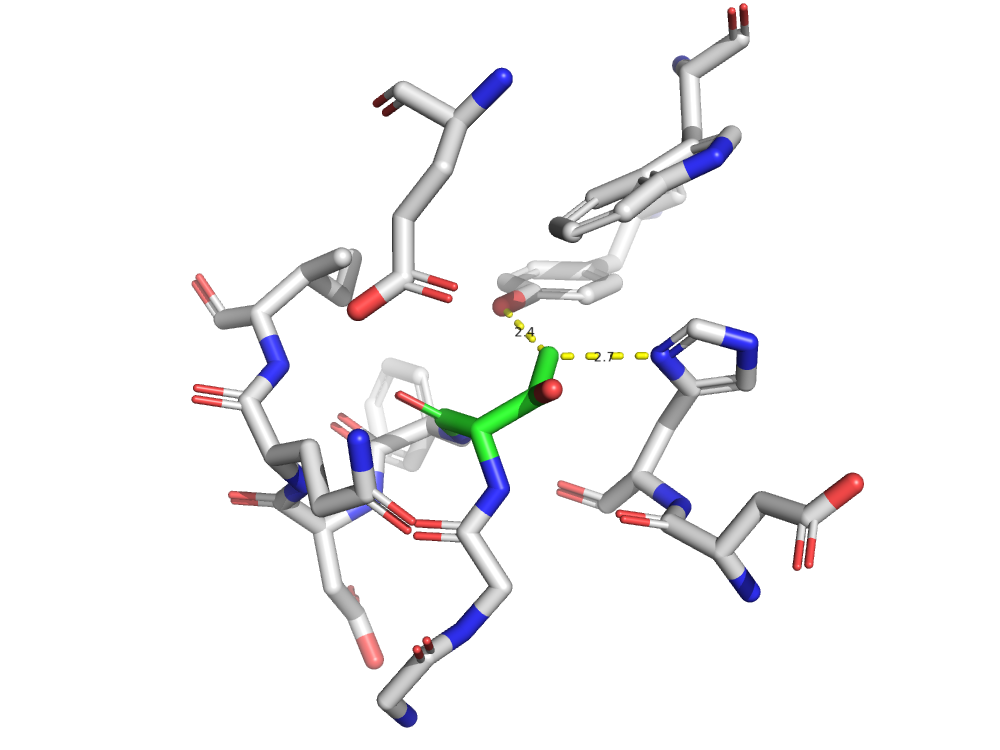
Все сообщество должно быть благодарным за то, что существует такой ресурс, как PDB Redo, ведь он способен предоставить структуры, в которых исправлены многие ошибки (refinement, rebuilding and validation). При поиске структуры 1SBP, в результатах можно сразу же заметить исправление остатка GLY`204, у которого была неправильная конформация главной цепи (рис. 11)
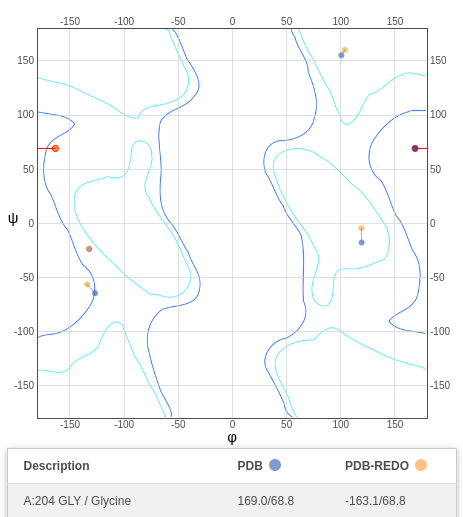
При сравнении двух структур (до и после), обнаруживаются изменения (рис. 12).
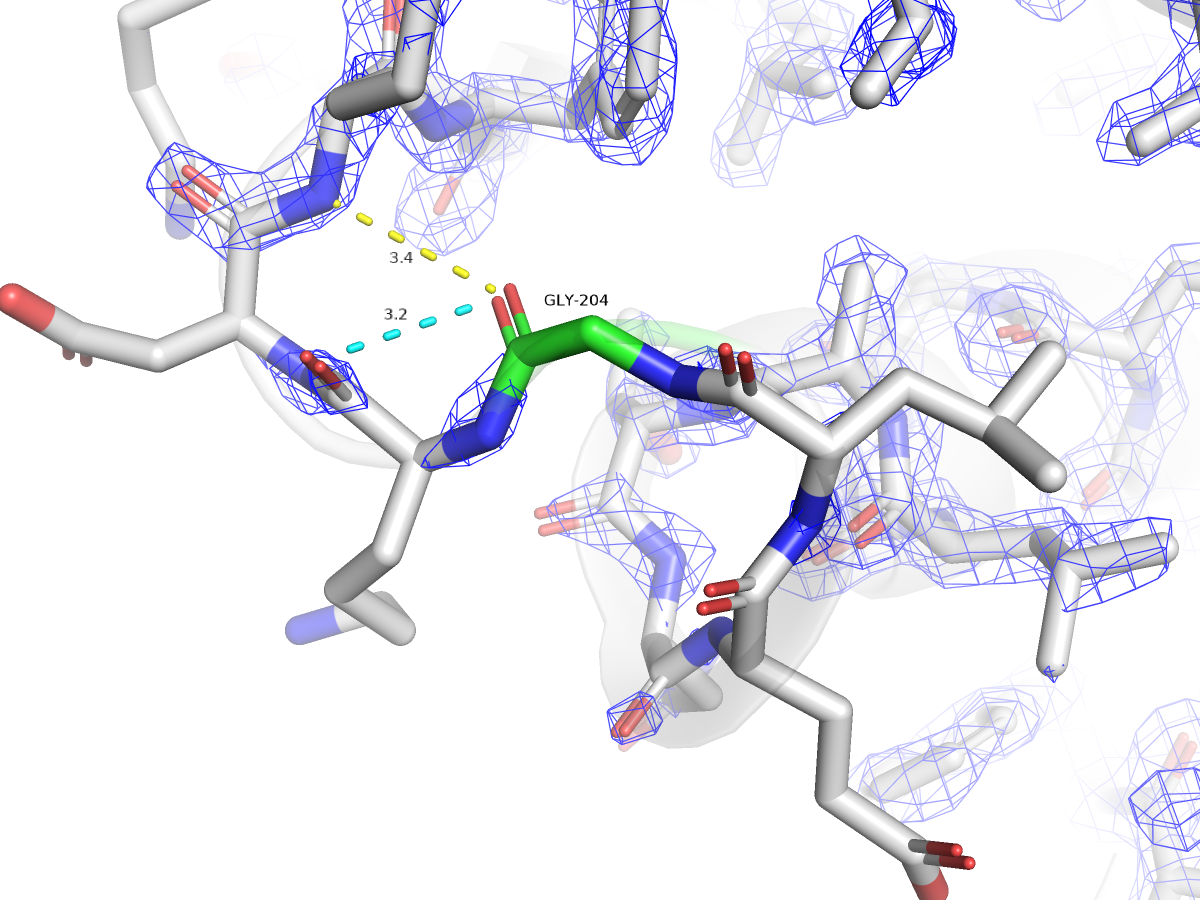
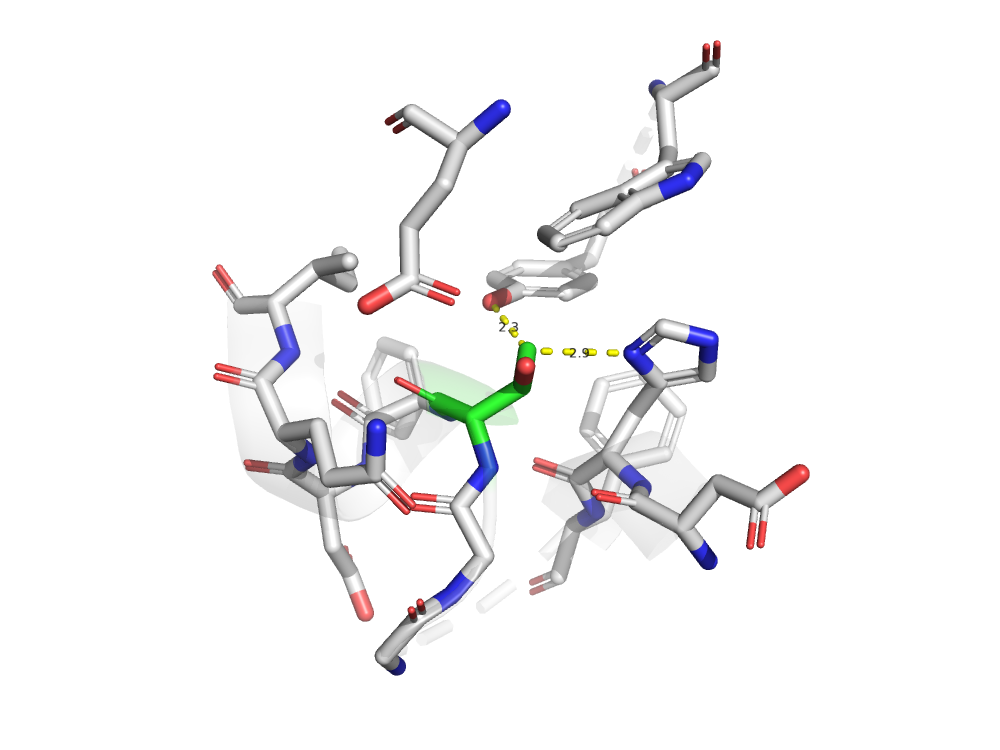
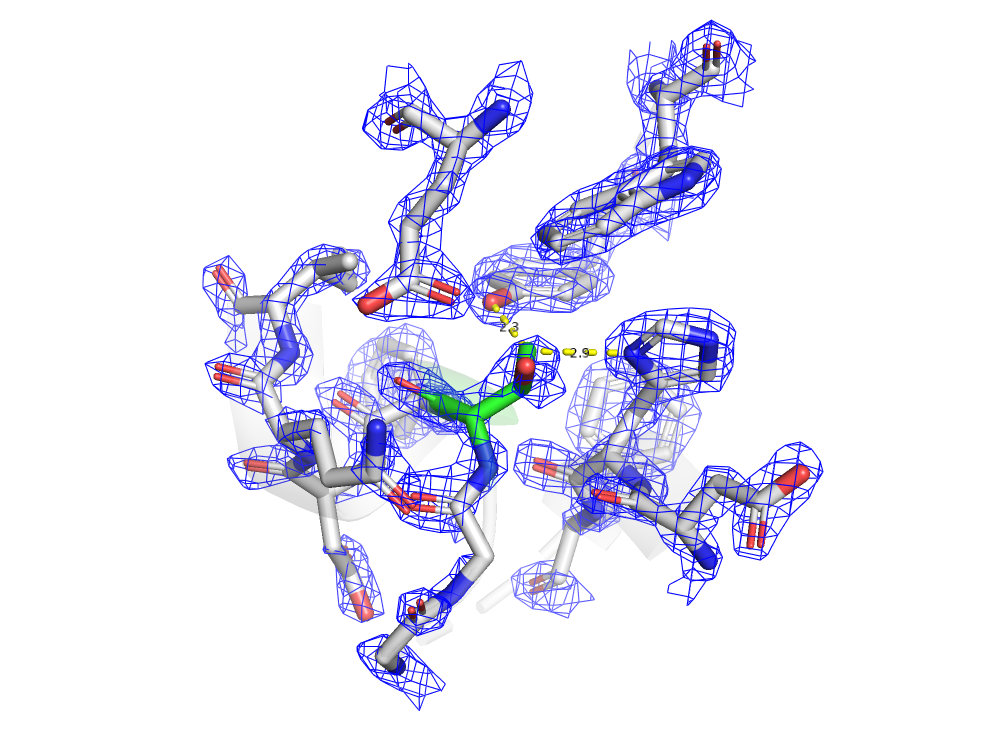
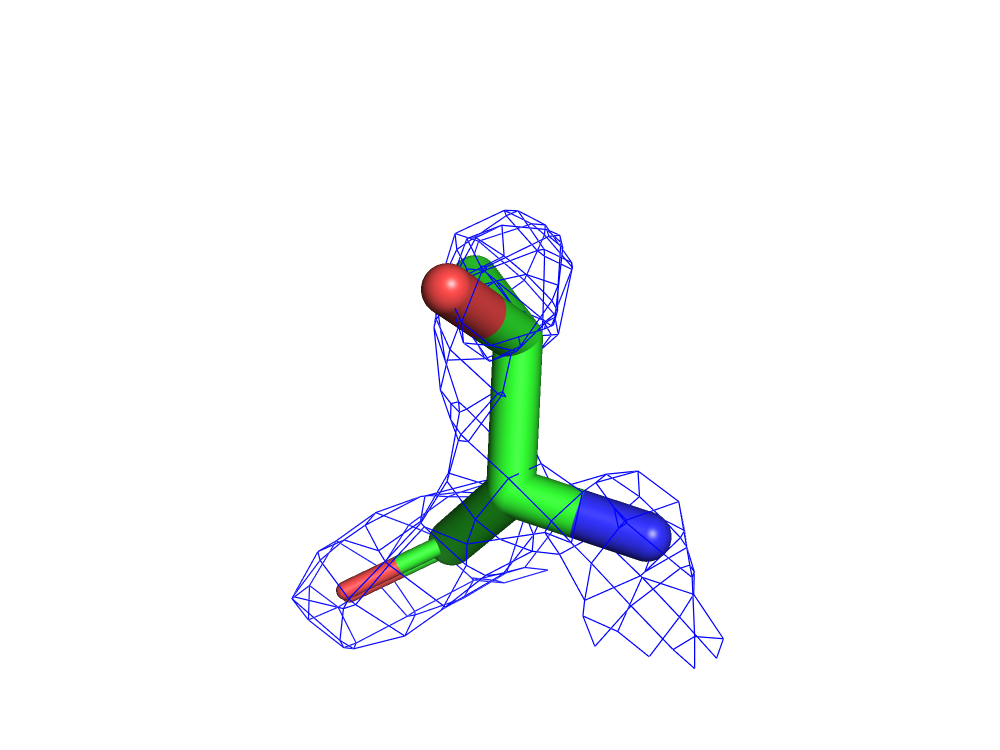
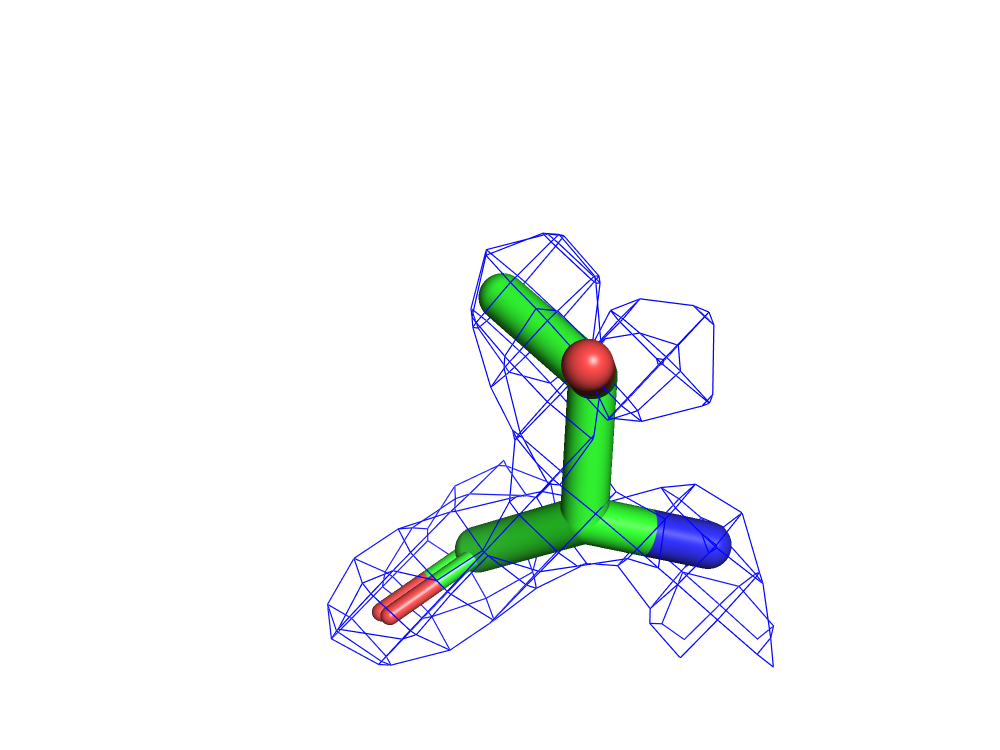
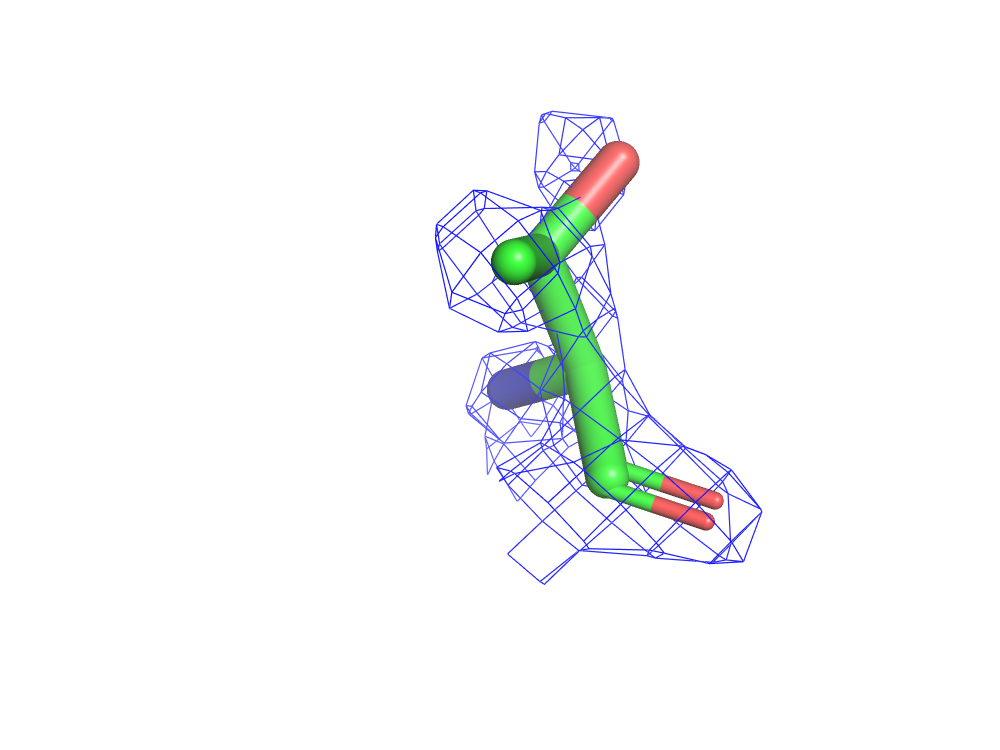
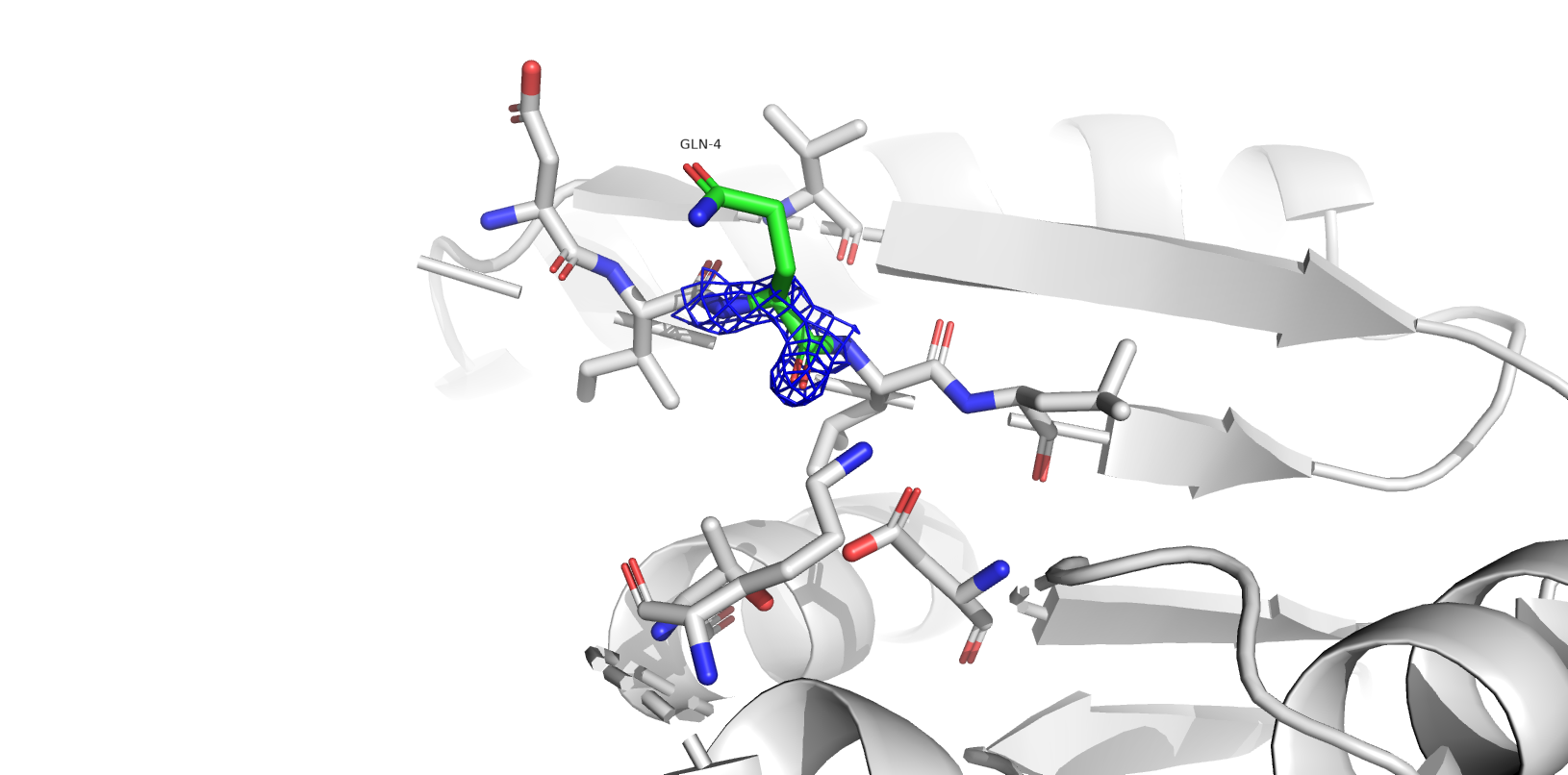
Сервис PDB Redo подтвердил, что все практически неточности структуры 1SBP из базы PDB связаны с ошибками расшифровки РСА, а не с особенностями структуры. При этом PDB Redo, как я думаю, не справился с уточнением конформации THR`303. Но и это, скорее всего, связано лишь с несовершенством алгоритмов расшифровки РСА, а не с особенностью структуры.
Список литературы
-
Финкельштейн А. В., Птицын О. Б
(2012)
Физика белка: курс лекций с цветными и стереоскопическими иллюстрациями и задачами.
3-е изд., испр. и доп. — М.: кдУ, 2012. — 456 с., [32] c. ил.: ил; -
Lovell SC, Davis IW, Arendall WB 3rd, de Bakker PI, Word JM, Prisant MG, Richardson JS, Richardson DC.
(2003)
Structure validation by Calpha geometry: phi,psi and Cbeta deviation.
Proteins. 15;50(3):437-50.