Практикум 8
Выбранный участок генома
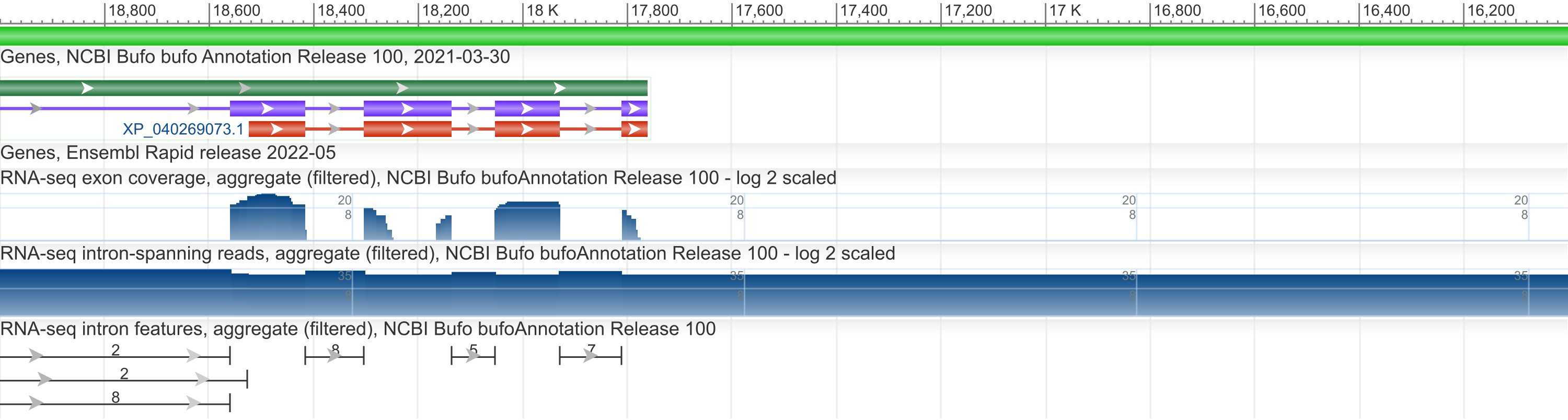
Я выбрала участок с 16001-го по 19000-й нуклеотид скэффолда NW_024400063 Unplaced Scaffold Reference aBufBuf1.1 (рис. 1). В нем содержится CDS XP_040269073.1, в которой аннотирован ген XlCGF66.1-подобного гаструльного белка с цинковыми пальцами (gastrula zinc finger protein XlCGF66.1-like). Выбранный участок можно скачать по ссылке.
Поправки критического значения E-value на множественные выравнивания
Математическое ожидание последовательностей, найденных по случайности, равно сумме E-value всех найденных последовательностей (из всех организмов). Поэтому нам необходимо вводить поправку критического значения, если мы хотим, чтобы матожидание количества найденных негомологических последовательностей (среди всех организмов) было меньше нужной границы. Я выбрала поправку Бонферрони и границу в 0,05, т.е. при n полученных результатов брала критическое значение в 0,05/n.
Это, конечно, не лучший подход, поскольку в таком случае от увеличения объема выдачи уменьшается количество значимых находок. Совсем по-честному надо бы ввести другую поправку.
Но если не делать никакую поправку и считать «значимыми» все находки BLAST, в какой-то момент матожидание случайно найденных выравниваний станет больше выбранной границы (и даже в какой-то момент больше 1).
Поиск BLAST в NCBI
Я искала гомологи моей последовательности в таксоне Vertebrata, но не в таксоне Hyloidea. Hyloidea — это надсемейство бесхвостых амфибий, в которое входит более половины их видов (из википедии). В таблице 1 я привожу информацию о результатах BLAST'a.
| Алгоритм | Таксон | Длина слова | Максимальное количество находок | Количество значимых находок | Количество значимых находок после поправки |
|---|---|---|---|---|---|
| megablast | Vertebrata, но не Hyloidea | 28 | 5000 | 109 | 108 |
| blastn | Vertebrata, но не Hyloidea | 11 | 5000 | 3688 | 2575 |
| blastx | Vertebrata, но не Hyloidea | 5 | 5000 | >=5000 | Не имеет смысла |
| tblastx | Vertebrata, но не Hyloidea | 3 | 5000 | Ошибка | Ошибка |
| tblastx | Reptilia | 3 | 500 | >=500 | Не имеет смысла |
Алгоритм megablast принимает на вход нуклеотидную последовательность и ищет сходство по базе нуклеотидных последовательностей. Его особенность в том, что длина слова у него очень большая, поэтому он работает быстро, но ищет только очень близкие гомологи (у которых есть длинные абсолютно одинаковые участки).
Алгоритм blastn принимает на вход нуклеотидную последовательность и ищет сходство по базе нуклеотидных последовательностей. У него уже короче слово, и он ищет менее близкие гомологи, но делает это медленнее, чем megablast.
Алгоритм blastx принимает на вход нуклеотидную последовательность, автоматически транслирует ее и ищет сходства получившихся «пептидов» по базам белковых последовательностей. Применим только для кодирующих белки последовательностей; более чувствительный, чем blastn.
Алгоритм tblastx принимает на вход нуклеотидную последовательность, автоматически транслирует ее и ищет сходства получившихся «пептидов» по базам так же транслированных нуклеотидных последовательностей. Он, наверное, может найти больше всего результатов, но работает дольше всех. Применим только для белок-кодирующих последовательностей.
Выравнивание рРНК E. coli на геном Bufo bufo
Я установила себе BLAST на персональный компьютер и проиндексировала геном Bufo bufo, используя следующую команду:
makeblastdb -in GCF_905171765.1_aBufBuf1.1_genomic.fna -dbtype nucl -out Bufo.fasta
После я нашла выровняла рРНК из файла на геном Bufo bufo:
blastn -task blastn -query rRNA_ecoli.txt -db Bufo.fasta -outfmt 7 > bbufo_ecoli_rRNA.txt
Результаты можно скачать по ссылке.
В рибосомах 16S рРНК нужна для инициации трансляции и для стабилизации правильных кодон-антикодоновых взаимодействий.
Я искала гомологи последовательностей 16S и 23S рРНК. Я искала их гомологи алгоритмом blastn, поскольку это последовательности, не кодирующие белок, и организмы, которые я сравниваю, слишком далеки друг от друга, чтобы использовать megablast. Я использовала параметры по умолчанию.
Для 16S нашлось 97 последовательностей (с поправкой 36 значимо), для 23S — 197 (125 значимых). Некоторые из них действительно выровнялись на последовательности, аннотированные в feature table как гены рРНК (например, участок в геноме 152610 — 157281, выровнявшийся на участок 23S рРНК E. coli, действительно находится в гене рРНК). Еще большое количество участков выровнялось почему-то не на сами гены рРНК, но очень рядом (в сотне-другой пар нуклеотидов). Почему так произошло, я пока не понимаю.