
Матрица BLOSUM62 (BLOcks SUbstitution Matrix) использует данные базы blocks и служит для выравнивания цитоплазматических белков. Число 62 в названии означает порог кластеризации, то есть все белки базы blocks идентичностью больше 62% объединяются в кластеры и рассматриваются как одно выравнивание. Матрица PHAt_T75_B73 же используется для выравнивания мембранных белков, поэтому ее значения отличаются от значений матрицы BLOSUM. Новая матрица была получена мной по таким же данным, как и те, что использовались для построения матрицы BLOSUM. Действительно, полученные значения очень мало отличаются.
| "аминокислота (1-буквенные обозначения)" | G | P | C | S | T | N | Q | D | E | H | R | K | A | M | I | L | V | F | W | Y |
| "аминокислота (3-буквенные обозначения)" | Gly | Pro | Cys | Ser | Thr | Asn | Gln | Asp | Glu | His | Arg | Lys | Ala | Met | Ile | Leu | Val | Phe | Trp | Tyr |
| BLOSUM62 | 6 | -2 | -3 | 0 | -2 | 0 | -2 | -1 | -2 | -2 | -2 | -2 | 0 | -3 | -4 | -4 | -3 | -3 | -2 | -3 |
| PHAT_T75_B73 | 9 | -3 | -2 | 1 | -1 | -1 | -2 | -2 | -3 | -4 | -5 | -4 | 1 | -1 | -2 | -2 | -2 | -2 | -5 | -3 |
| Реконструкция | 5 | -2 | -2 | -1 | -2 | -1 | -2 | -2 | -2 | -2 | -2 | -2 | 0 | -3 | -4 | -4 | -3 | -3 | -3 | -3 |
Таблица 1. Сравнение матриц аминокислотных замен (BLOSUM62, PHAT_T75_B73 и реконструированной на основании базы blocks) на примере аминокислоты глицина.
Для белка YPJD_BACSU скриптом evolve_protein.pl были получены несколько коротких фрагментов (по 20 аминокислотных остатков) из искусственно смоделированного мутанта YPJD_BACSU. Скрипт evolve_protein.pl создает эволюционно измененную последовательность белка с учетом наследственности и изменчивости, но без учета естественного отбора.
Параметр этого скрипта –c ($def_change) отвечает за замену аминокислотного остатка на другой (для каждой аминокислоты замена на любую другую, в том числе и на саму себя, происходит с вероятностью 1/20), либо удаление этой аминокислоты из последовательности, либо вставку новой аминокислоты. Значение –c=0.6 соответствует тому, что в 60 % аминокислотных остатков будет происходить замена. Параметр –d ($def_replace) показывает, с какой вероятностью будет происходить замена аминокислотного остатка, если его позиция уже была изменена. Если замены не происходит, то с равной вероятностью происходит делеция или инсерция.
Алгоритмы Нидлмана-Вунша и Смита-Ватермана выполняются программами из пакета EMBOSS needle и water, соответственно. При этом результатом применения алгоритма Нидлмана-Вунша является выравнивание по наибольше по длине последовательности, то есть получается полноразмерное выравнивание, тогда, как алгоритм Смита-Ватермана находит наибольшие по весу локальные выравнивания, и иногда такие выравнивания имеют мало общего с ходом эволюции белка. Чем меньше последовательность, тем больше вероятность, что в большей последовательности попадется участок, случайно совпадающий с этой последовательностью. И для того, и для другого алгоритма можно задать следующие параметры: штраф за гэп (первый - открывающий, по умолчанию = 10) и штраф за последующие гэпы (0.5).
| № мут. | Механизм выравнивания | Выравнивание | Identity,% | Similarity,% | Score |
| 1 | Вручную | 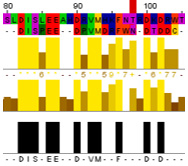 |
48 (по участку) или 10 (по всему выравниваю) | 57 или 12 | 25 |
| Нидлман-Вунш |  |
10 | 12 | 27 | |
| Смит-Ватерман |  |
60 | 67 | 31 | |
| 2 | Вручную | 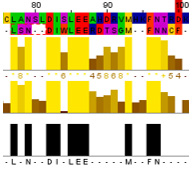 |
42 или 9 | 58 или 13 | 16 |
| Нидлман-Вунш |  |
9 | 10 | 15 | |
| Смит-Ватерман |  |
75 | 75 | 22 | |
| 3 | Вручную |  |
61 или 12 | 65 или 13 | 53 |
| Нидлман-Вунш | 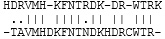 |
12 | 12 | 50 | |
| Смит-Ватерман |  |
78 | 78 | 52 |
Таблица 2. Сравнение выравниваний, полученных различными способами.
Значения identity, similarity и вес по матрице BLOSUM62 (score), полученные вручную, очень близки к значениям, полученным при использовании алгоритма Нидлмана-Вунша. А алгоритм Смита-Ватермана выдает значения для более коротких последовательностей (короче, чем по участкам, для которых значения были посчитаны вручную, но обязательно учитывали всю последовательность мутантного белка), чем и обоснованы очень высокие значения.
| Дата последнего изменения: 26/04/2013. |
|
© Trushina Nataliya |