
Нужно построить филогенетическое дерево тех же бактерий, что в предыдущих заданиях (Таблица 1), используя последовательности РНК малой субъединицы рибосомы (16S rRNA).
Таблица 1. Интересующие нас параметры для ранее отобранные бактерий.
| Название | Мнемоника | AC записи EMBL | Координаты 16S rRNA | Цепь (прямая/обратная) |
| Bacillus subtilis | BACSU | AL009126 | 96392..97945 | прямая |
| Clostridium tetani | CLOTE | AE015927 | 41801..43309 | обратная |
| Enterococcus faecalis | ENTFA | AE016830 | 2771880..2773401 | обратная |
| Finegoldia magna | FINM2 | AP008971 | 197837..199361 | прямая |
| Geobacillus kaustophilus | GEOKA | BA000043 | 10421..11973 | прямая |
| Staphylococcus aureus | STAA1 | AP009324 | 576283..577837 | прямая |
| Streptococcus pyogenes | STRP1 | AE004092 | 264453..265788 | прямая |
| Listeria monocytogenes | LISMO | CP007492.1 | 242732..244284 | прямая |
С помощью NCBI-Nucleotide и команды "seqret embl:* -sask" получили последовательности, кодирующие 16S rRNA.
Алгоритмом Muscle произвели выравнивание в JalView.
Ссылка на выравнивание.
По этому выравниванию в программе MEGA по алгоритму Neighbour-Joining построили приведенное ниже дерево.

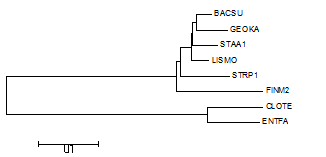
Рисунок 1. Правильное дерево (слева) и дерево, построенное по последовательностям 16S rRNA методом Neighbour joining (справа).
Реконструированное дерево довольно сильно отличается от правильного. Хотя есть одинаковые ветви: (BACSU, GEOKA, LISMO, STAA1) vs (CLOTE, FINM2, ENTFA, STRP1), BACSU, GEOKA) vs (CLOTE, FINM2, ENTFA, STRP1, LISMO, STAA1).
Так как новое дерево строили по нуклеотидным последовательностям, можно было предположить, что оно будет хуже настоящего и других - построенных по белковым (4 буквы - больше вероятность ошибки, чем при 20). А также участки 16s РНК выбирались случайно.
Сравнение реконструкций по белкам и нуклеотидным последователям.
В новом дереве две совпадающие с оригинальным деревом ветви, что почти совпадает по количеству с реконструкциями по белкам (в предыдущих реконструкциях совпадали 1-3 ветви).
Ищу в выбранных мной бактериях достоверные гомологи белка CLPX_BACSU.
Для этого использую файл proteo.fasta на диске P (там лежат записи банка UNIPROT бактерий из задания 1) Произвожу поиск гомологов программой blastp с порогом на E-value равным 1e-5. По мнемонике видов отбираю только те находки, которые относятся к выбранным мной бактериям. Произвожу множественное выравнивание с помощью Muscle with defaults.

Рисунок 2. Множественное выравнивание найденных гомологов.
Строю дерево этих гомологов с помощью программы MEGA по алгоритму по алгоритму Neighbor-Joining с поддержкой Bootstrap - 500 реплик. Считаю дерево реконструированным верно. Два гомологичных белка называем ортологами, если они а) из разных организмов; б) разделение их общего предка на линии, ведущие к ним, произошло в результате видообразования. Два гомологичных белка из одного организма называем паралогами.
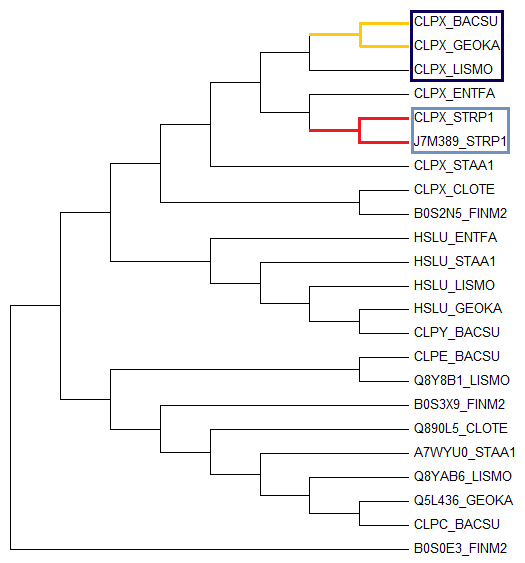
Рисунок 3. На дереве темно-синим квадратом обозначены ортологи, голубым - паралоги; а также приведены примеры отражённых на дереве эволюционных событий двух типов: 1) красные ветви - дупликация гена, 2) желтые - разделение путей эволюции белков в результате видообразования.
| Дата последнего изменения: 14/09/2013. |
|
© Trushina Nataliya |