11. Пространственное выравнивание и совмещение
1. Построение совмещений в PyMOL
Необходимо построить три совмещения цепи А записи 1HY0 и цепи А записи 1I0A (по каждому из доменов CATH). Для этого скачаем с сайта PDB записи 1HY0.pdb и 1I0A.pdb, а также при помощи сервера pDomains посмотрим, как цепи А этих записей разделяются на домены методом CATH:1HYO, chain A: 1HY0A1 27-123, 195-234 1HY0A2 124-194, 235-364, 437-463 1HY0A3 365-436 1I0A, chain A: 1I0AA1 27-123 , 195-234 1I0AA2 124-194 , 235-364 , 437-463 1I0AA3 365-436Далее построим совмещения по каждому из доменов при помощи команды в PyMOL:
align <домен1>,<домен2>На всех изображениях совмещений красным показан участок структуры 1HY0, зелёным - 1I0A.
Совмещение первых доменов: RMSD = 0,371
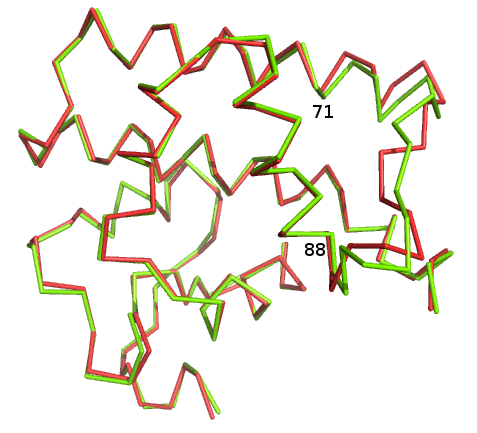
Видно, что домены хорошо совпадают, за исключением участка 71-88.
Совмещение вторых доменов: RMSD = 0,409
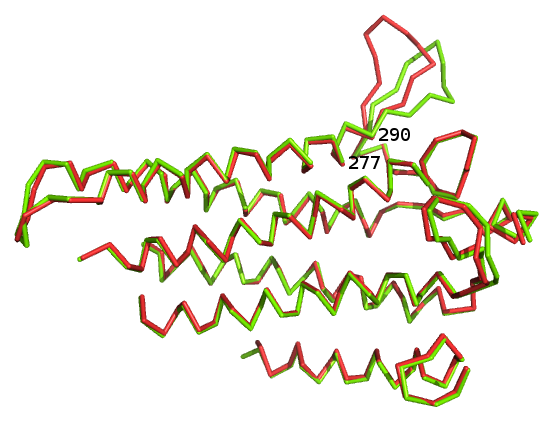
Здесь домены также совпадают практически полностью. Из общей картины выбивается только "петля" между остатками 277-290.
Совмещение третьих доменов: RMSD = 0,527
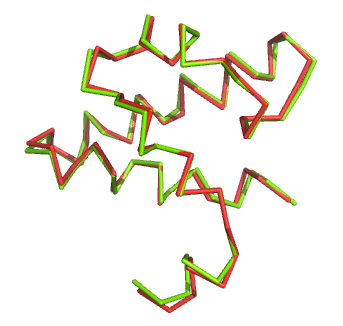
В этом случае домены совместились практически идеально.
2. Построение структурного выравнивания
При помощи программы PDBeFOLD (она же SSM) построим структурное выравнивание цепи A из 1AKH и цепи A из 1W0T. Затем по данному выравниванию с помощью сервиса Geometrical core найдём геометрическое ядро с порогом 2Å. Для этого необходимо изменить названия последовательностей. В результате получаем таблицу аминокислотных остатков, образующих геометрическое ядро:| Pos. | 1AKH_A | 1W0T_A |
| 11 | ALA83 | LYS389 |
| 12 | PHE84 | ASN390 |
| 13 | LEU85 | LEU391 |
| 15 | GLU87 | SER393 |
| 28 | LYS100 | TRP403 |
| 29 | GLU101 | SER404 |
| 30 | GLU102 | LYS405 |
| 31 | VAL103 | ILE406 |
| 41 | THR110 | THR416 |
| 42 | PRO111 | SER417 |
| 43 | LEU112 | VAL418 |
| 44 | GLN113 | MET419 |
| 45 | VAL114 | LEU420 |
| 46 | ARG115 | LYS421 |
| 47 | VAL116 | ASP422 |
| 48 | TRP117 | ARG423 |
| 49 | PHE118 | TRP424 |
| 50 | ILE119 | ARG425 |
| 51 | ASN120 | THR426 |
| 52 | LYS121 | MET427 |
| 53 | ARG122 | LYS428 |
| 54 | MET123 | LYS429 |
| 55 | ARG124 | LEU430 |
По СА-атомам, входящим в геометрическое ядро, совместим структуры в PyMOL командой pair_fit:
pair_fit 1AKH and (resi 83-85,87,100-103,110-124) and chain a and name ca,
1W0T and (resi 389-391,393,403-406,416-430) and chain a and name ca
RMSD = 0,865В результате получено следующее изображение геометрического ядра (здесь и далее структура 1AKH выделена зелёным, 1W0T - сиреневым):
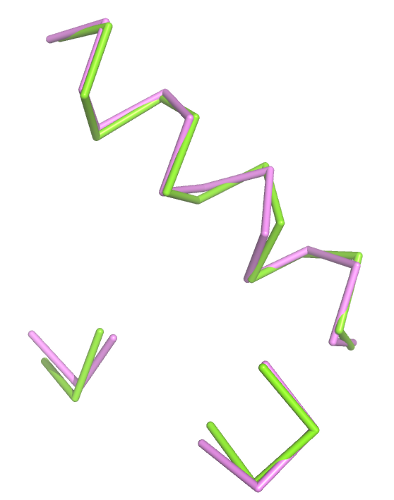
Полные цепи, совмещённые по геометрическому ядру:

Совмещение довольно неплохое, о чём свидетельствует и значение RMSD.
Теперь совместим полные цепи командой align:
align 1AKH & chain a & name ca,1W0T & chain a & name caRMSD = 3,231
Видно, что цепи совместились совершенно неправильно:
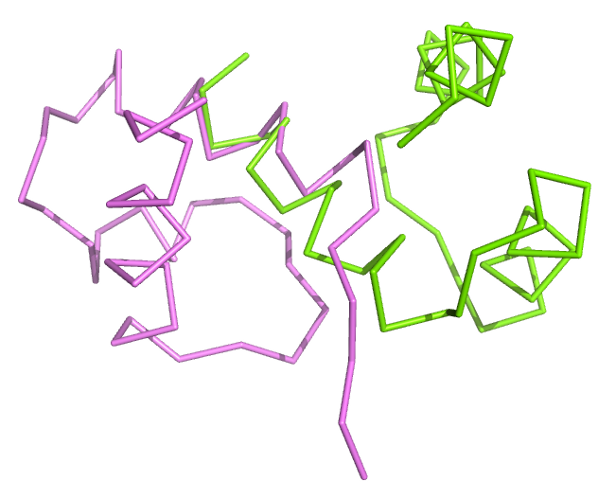
Можно совместить цепи не только по СА-атомам:
align 1AKH & chain a,1W0T & chain aRMSD = 1,340

В этом случае значение RMSD ниже, чем в предыдущем, да и цепи совместились качественнее. Но совмещёнными остались только те же небольшие участки цепей, что и при совмещении по СА-атомам.
Теперь рассмотрим, как работает команда super:
align 1AKH & chain a,1W0T & chain aRMSD = 2,186
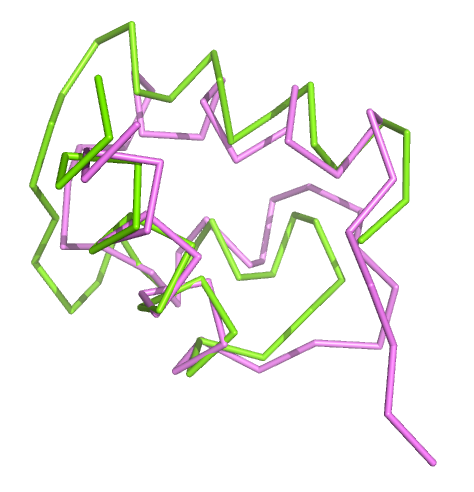
Значение RMSD для такого совмещения не особенно хорошее, но из рисунка видно, что цепи совместились куда лучше, чем в случае команды align.
Назад