.jpg)
Для начала я скопировала в дирректорию /nfs/srv/databases/ngs/nknjasewa файлы с ридами (файлы с одноконцевыми чтениями, chr6.fastq) и хромосомой (хромосомы человеческого генома (сборка версии hg19), chr6.fasta). Все выполненные команды приведены в таблице 2.
Я сделала контроль качества чтений с помощью программы FastQC. В результате работы программы я получила архив (chr6_fastqc.zip), который содержит отчет о программе в виде html файла. Последовательностей с плохим качеством обнаружено не было. Общее число последовательностей - 10289. Длина послевательностей 33-100. GC состав - 40%. На рис.1 изображение "Per base sequence quality", полученное с помощью FastQC до чистки.
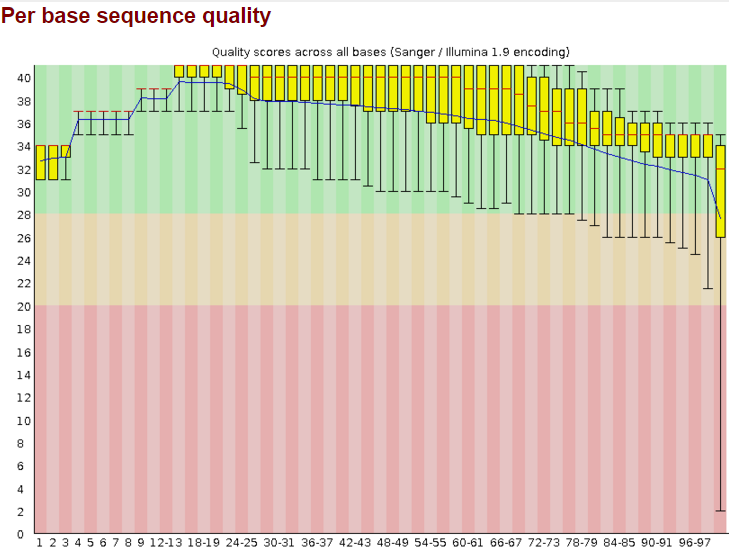
Рис.1 Качество чтений до чистки
Затем я сделала очистку чтений с помощью программы Trimmomatic. Так как в чтениях адаптеры уже удалены, я отрезала с конца каждого чтения нуклеотиды с качеством ниже 20, оставила только чтения длиной не меньше 50 нуклеотидов. После этого я снова проверила качество чтений программой FastQC. На рис.2 изображение "Per base sequence quality", полученное с помощью FastQC после чистки. Общее число последовательностей - 10120. Длина послевательностей 50-100. Полученные данные свидетельствуют о том, что качество чтений значительно улучшилось. Были удалены концы с качеством ниже 20 и длина оставшихся чтений не менее 50.
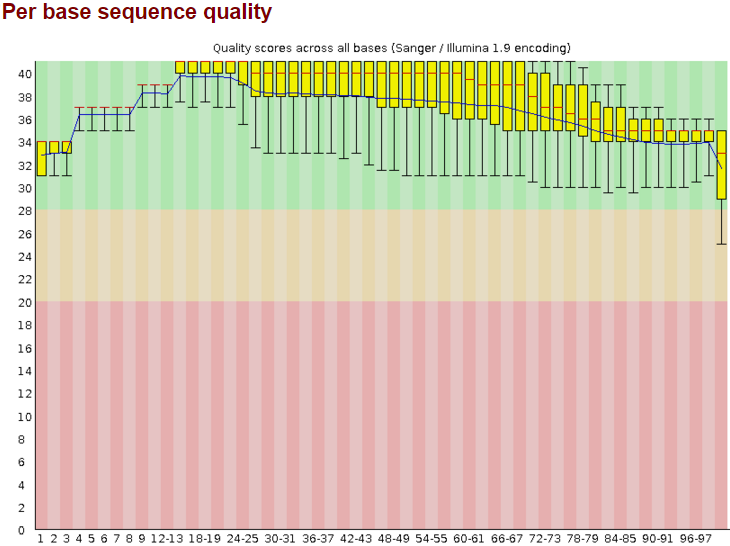
Рис.2 Качество чтений после чистки
Картирование чтенийДля начала я отсканировала чтения с помощью программы BWA (руководство). Для этого было необходимо проиндексировать референсную последовательность 6-ой , а затем построить выравнивание прочтений и референса в формате .sam.
Для анализа выравнивания я перевела выравнивание чтений с референсом в бинарный формат .bam, используя пакет samtools, команда view. Затем я отсортировала выравнивание чтений с референсом (получившийся после картирования .bam файл) по координате в референсе начала чтения, команда sort. После этого я проиндексировала отсортированный .bam файл командой index. Командой idxstats я узнала, сколько чтений откартировалось на геном. На рис.3 приведена полученная данной командой таблица. Видно, что все 10120 чтений откартировались на референсную последовательность 6-ой хромосомы (10120 - число чтений, картированных на хромосому, 0 - число чтений, не картированных на хромосому), длина последовательности - 171115067.
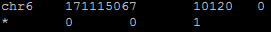
Рис.3 Результат работы команды idxstats пакета samtools
Анализ SNPДля осуществления поиска SNP и инделей потребовалось выполнить следующие действия. Для начала я создала файл с полиморфизмами в формат е .bcf с помощью команды mpileup пакета samtools с опцией -uf. Затем я создала файл со списком отличий между референсом и чтениями в формате .vcf, используя для этого команду call пакета bcftools. Полученный файл: plm.vcf. Количество инделей: 11. Количество SNP (отличий последовательности ДНК размером в один нуклеотид): 83. Больше чем у половины найденных полиморфизмов покрытие очень хорошее (больше 20), примерно у трети найденных полиморфизмов покрытие плохое (от 1 до 2). Качество найденных полиморфизмов соответсвтует покрытию.
Таблица 1. Описание трех полиморфизмов
| № полиморфизма | Координата | Тип полиморфизма | В референсе | В чтениях | Глубина покрытия | Качество чтений |
| 1 | 106989212 | Однонуклеотидная замена | G | A | 36 | 221.999 |
| 2 | 154357986 | Однонуклеотидная замена | T | G | 1 | 11.3429 |
| 3 | 107016838 | Делеция (индель) | CTTT | CTTTTT | 35 | 212.468 |
С помощью программы annovar я проаннотировала полученные snp, используя базы данных: refgene, dbsnp, 1000 genomes, GWAS, Clinvar.
SNP при аннотации по базе данных RefGene классифицируются по положению, которое они занимают в геноме, то есть попадают ли они в экзоны или интроны генов, в межгенные области, в гены некодирующих РНК или в 3'- и 5'-некодирующие области (UTR3 и UTR5). В моем случае в экзоны попадает 15, в интроны 65, в межгенные области 3 snp.
Для выявления SNP, имеющих rs, использовался скрипт annotate_variation.pl. Получено два файла: dropped - snp, имеющие rs, то есть те, которые аннотированы в dbSNP (для таких SNP указываеся их идентификатор (rs) в данной БД) - в моем случае таких snp 77; filtered - snp без rs (неаннотированые в dbSNP) - в моем случае таких snp 6.
Описание нуклеотидных и аминокислотных замен представлено на рис. 4.
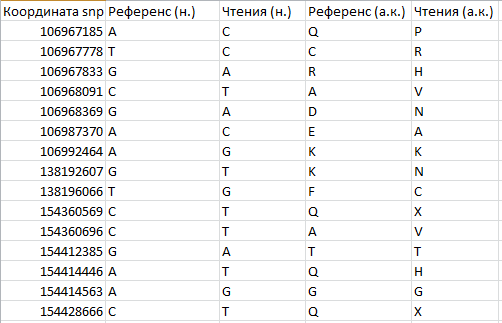
Рис.4 Нуклеотидные и аминокислотные замены
Таблица 2. Выполненные команды
| Команда | Описание |
| fastqc chr6.fastq | Выдает архив, который содержит отчет о программе в виде html файла. С помощью данной программы можно проверить качество чтений. |
| java -jar /usr/share/java/trimmomatic.jar SE -phred33 chr6.fastq chr6_out.fastq LEADING:20 TRAILING:20 MINLEN:50 | LEADING: отрезает нуклеотиды от начала каждого чтения, если ниже порогового значения. TRAILING: отрезает нуклеотиды от конца каждого чтения, если ниже порогового значения. MINLEN: перестает читать, если чтение ниже заданной длины. |
| bwa index chr6.fasta | Индексирует референсную последовательность. |
| bwa mem chr6.fasta chr6_out.fastq > aln-se.sam | Строит выравнивания прочтений и референса в формате .sam. |
| samtools view aln-se.sam -b -o aln-se.bam | Переводит выравнивание чтений с референсом в бинарный формат .bam. -b - задает формат выдачи bam (бинарный); -o - задает имя файла выдачи. |
| samtools sort -T out.prefix -o aln-sort.bam aln-se.bam | Сортирует выравнивание чтений с референсом (получившийся после картирования aln-se.bam файл) по координате в референсе начала чтения. -o - запись вывода в файл, а не в стандартный вывод; -T - необходимая опция записи временных файлов |
| samtools index aln-sort.bam | Индексирует отсортированный файл aln-sort.bam. |
| samtools idxstats aln-sort.bam | Показывает, сколько чтений откартировалось на геном. |
| samtools mpileup -uf chr6.fasta aln-sort.bam -o plm.bsf | Создает файл с полиморфизмами в формате .bcf. -о - запись вывода в файл, а не в стандартный вывод; -f - файл с референсной последовательностью; -u - создание несжатого файла с выходом. |
| bcftools call -cv -o plm.vcf plm.bsf | Создает файл со списком отличий между референсом и чтениями в формате .vcf |
| grep 'INDEL;' plm.vcf | wc -l | Считает количество инделей |
| grep 'DP=' plm.vcf | wc -l | Считает количество полиморфизмов, по разности с предыдущим можно узнать количество snp |
| perl /nfs/srv/databases/annovar/convert2annovar.pl plm.vcf > snp.human | Данный скрипт преобразует формат .vcf файла в формат .vcf4, с которым умеет работать программа annovar. |
| perl /nfs/srv/databases/annovar/annotate_variation.pl -out ann_refgene -build hg19 snp.human /nfs/srv/databases/annovar/humandb/ | Аннотирует по базе данных refgene |
| perl /nfs/srv/databases/annovar/annotate_variation.pl snp.human -build hg19 /nfs/srv/databases/annovar/humandb/ -filter -dbtype snp138 -out ann_dbsnp | Аннотирует по базе данных dbsnp |
| perl /nfs/srv/databases/annovar/annotate_variation.pl snp.human -filter -buildver hg19 /nfs/srv/databases/annovar/humandb/ -dbtype 1000g2014oct_all -out ann_1000g | Аннотирует по базе данных 1000 genomes |
| perl /nfs/srv/databases/annovar/annotate_variation.pl -regionanno snp.human -build hg19 -dbtype gwasCatalog /nfs/srv/databases/annovar/humandb/ -out ann_GWAS | Аннотирует по базе данных GWAS |
| perl /nfs/srv/databases/annovar/annotate_variation.pl snp.human /nfs/srv/databases/annovar/humandb/ -filter -dbtype clinvar_20150629 -buildver hg19 -out ann_Clinvar | Аннотирует по базе данных Clinvar |