.jpg)
2. Один файл в формате fasta с несколькими последовательностями разделить на отдельные fasta файлы.
Команда: seqretsplit -seq coding1.fasta seq.fasta.
В результате были созданы отдельные файлы, каждый из которых содержит ровно одну последовательность
из файла coding1.fasta.
3. Из файла с хромосомой в формате .gb вырезать три кодирующих последовательности по указанным
координатам "от", "до", "ориентация" и сохранить в одном fasta файле.
Команда: seqret @CDS.fasta.
Из файла chromosome.gb я извлекла AC (U00096) и скачала fasta файл из базы данных GenBank с последовательностью
хромосомы (sequence.fasta).
Я выбрала три кодирующие последовательности из файла chromosome.gb и записала координаты этих
последовательностей в файл CDS.fasta в таком виде:
sequence.fasta[190:255]
sequence.fasta[337:2799]
sequence.fasta[9928:10494:r]
Затем я ввела указанную команду и получила файл
u00096.fasta, в котором содержатся три нужных последоватльности.
4. Транслировать кодирующие последовательности, лежащие в одном fasta файле, в аминокислотные,
используя указанную таблицу генетического кода. Результат - в одном fasta файле.
Команда: transeq coding1.fasta prot.fasta.
Я взяла файл coding1.fasta. Для трансляции используется
стандартная таблица генетического кода и рамка без сдвига, так как в данном файле все кодирующие
последовательсти начинаются с ATG. В результате был создан файл
prot.fasta, который
содержит аминокислотные последовательности.
5. Транслировать данную нуклеотидную последовательность в шести рамках.
Команда: transeq coding.fasta prot_frame.fasta -frame 6.
Я взяла файл coding1.fasta. Для трансляции используется
стандартная таблица генетического кода. В результате был создан файл
prot_frame.fasta, который
содержит аминокислотные последовательности, полученные из всех рамок считывания.
6. Перевести выравнивание и из fasta формата в формат .msf.
Команда: seqret align.fasta msf::align.msf.
Я взяла файл align.fasta. В результате был создан файл
align.msf в формате msf.
7. Выдать в выходной поток число совпадающих букв между второй последовательностью выравнивания
и всеми остальными (на выходе только имя последовательности и число).
Команда: infoalign all_align.fasta -refseq 2 -only -name -idcount all_align.infoalign.
Я взяла файл all_align.fasta. В результате был создан файл
all_align.infoalign.
Я выбрала бактерию Cyanothece sp. PCC 8801, которая содержит одну хромосому (идентификатор NCBI: NC_011726) и три плазмиды. Из базы данных NCBI я скачала аннотацию в формате genbank (sequence.gb) и последовательность в формате fasta (sequence.fasta). Для получения трансляции открытых рамок я использовала команду getorf пакета EMBOSS. Команда: getorf sequence.fasta -table 11 -minsize 180 -circular -find 0 ORFs.fasta. Полученный файл: ORFs.fasta. Значения опций указаны в таблице 1.
Таблица 1. Опции команды getorf.
| Опция | Значаение |
| -table 11 | Таблица генетического кода для генома бактерии |
| -minsize 180 | Минимальная длина открытой рамки - 180 п.н. |
| -circular | Кольцевая хромосома |
| -find 0 | Выходные последовательности - трансляции открытых рамок от стоп кодона до стоп кодона |
Для получения списока координат и ориентаций найденных открытых рамок я использовала infoseq. Команда: infoseq ORFs.fasta -only -name -length -description -outfile out.txt. Значения опций указаны в таблице 2.
Таблица 2. Опции команды infoseq.
| Опция | Значаение |
| -only | Показывать только указанные параметры |
| -name | ID открытой рамки |
| -length | Длина трансляции в остатках |
| -description | Описание содержит координаты открытых рамок |
Обработанные результаты работы команды infoseq представлены в файле ORFs.xlsx.
Для получения списка аннотированных генов белков я скачала файлы с расширениями .ptt (хромосомная таблица со списком генов белков)и .faa (с последовательностями белков в формате fasta): NC_011726.ptt, NC_011726.faa. Обработав файл NC_011726.ptt я получила таблицу аннотированных генов белков в формате Excel: ann.xlsx.
Для сравнения двух полученных таблиц я использовала Excel. Я добавила в каждую таблицу первый столбец source и написала в нем ORF в первой таблице и Annotation - во второй. Затем я объединила обе таблицы в одну и отсортировала по "from". Полученная таблица: res.xlsx.
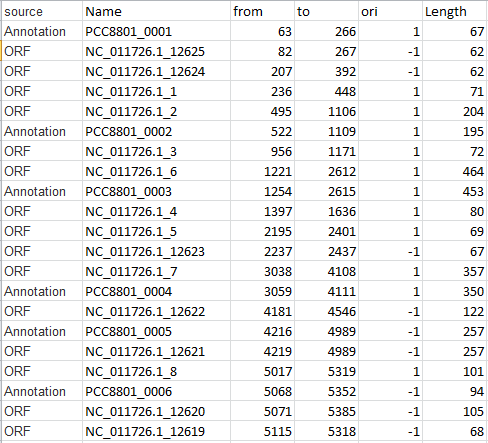
Рис.1 Фрагмент таблицы сравнения аннотированых и не аннотированных генов белков.
Из таблицы видно, что аннотированных белков примерно в три раза больше, чем не аннотированных. Координаты конца аннотированного гена в среднем на три нуклеотида дальше, чем у аннотированного. Координаты начала не аннотрированного гена довольно сильно отличаются от координат начала не аннотированного гена, возможно, это из-за того, что я использовала трансляцию открытых рамок от стоп кодона до стоп кодона.