.jpg)
Системы рестрикции-модификации (Р-М) - это один из механизмов защиты прокариот от чужеродной ДНК, например, бактериофагов. Система Р-М умеет распознавать определенные короткие последовательности ДНК (сайты рестрикции) и гидролизовать ДНК, если эти последовательности не метилированы. В ДНК клетки все сайты рестрикции заметилированы, а в ДНК фагов - нет. Поэтому клеточная ДНК остается невредимой, а ДНК фагов гидролизуется. Но иногда в процессе метилирования сайтов рестрикции случаются ошибки и геном бактерии может быть гидролизован. Из-за этого бактерии выгодно содержать в геноме как можно меньше сайтов узнавания систем Р-М, чтобы уменьшить вероятность случайного гидролиза.
Отбор против сайтов в геноме можно обнаружить, сравнив наблюдаемое число сайтов с ожидаемым числом. Например, можно вычислить отношение наблюдаемое/ожидаемое число сайтов (контраст), и если это отношение меньше 1, значит встретилось меньше сайтов, чем ожидалось. Обнаружив такие "избегаемые" сайты можно предсказать, какие системы Р-М бактерия содержит (или содержала в недавнем прошлом).
Бактерии достаточно быстро меняют набор систем Р-М. Поэтому даже бактерии (или археи) одного вида могут содержать разный набор систем Р-М в разных популяциях. Цель данного задания состоит в том, чтобы сравнить предполагаемые (по избеганию сайтов) наборы систем Р-М в полном геноме бактерии из NCBI и наборе контигов того же вида из метагенома кишечника человека.
Бактерия, в которой я искала системы Р-М, — Eggerthella lenta (GenBank AC: CP001726.1). Файл, содаржащий список почти всех известных сайтов систем Р-М типа II (основной тип), — sites.txt.
С помощью веб-сервиса было посчитано ожидаемое количество и контраст всех сайтов из списка в геноме названной бактерии, используя метод Карлина. Выходной файл веб-сервиса: tmpMwr26l.tsv. Затем были отобраны все сайты, для которых контраст не более 0.78 (это порог, чтобы отличие от 1 можно было считать значительным): b.tsv. Количество таких сайтов: 2.
Был осуществлен поиск избегаемых сайтов рестрикции в наборе контигов из метагенома кишечника человека. Выходной файл веб-сервиса: tmpiqJuCk.tsv. Затем были отобраны все сайты, для которых контраст не более 0.78 (это порог, чтобы отличие от 1 можно было считать значительным): m.tsv. Количество таких сайтов: 2.
Оба штамма имеют всего по две системы Р-М, однако только один избегаемый сайт CTAG был найден как в полном геноме бактерии, как и в контигах из метагенома кишечника человека. Бактерия в кишечнике человека более защищена от окружающей среды, поэтому, возможно, имеет меньше Р-М систем. Но делать какие-либо выводы на основании полуенных данных я не могу, так как было найдено равное количество P-M сайтов.
Нахождение последовательностей Шайн-Дальгарно в геноме бактерии или археи, данном вам в первом семестре.Последовательность Шайна-Дальгарно (SD) - участок связывания малой субъединицы прокариотической рибосомы с мРНК, обычно расположенный за несколько нуклеотидов до начала трансляции. Считается, что у прокариот SD присутствует в начале большинства генов, и мутации в ее консенсусной последовательности GGAGG или в комплементарном ей участке 16s рРНК приводят к подавлению трансляции. По данным статей, эта последовательность в общем случае составляет 5-6 нуклеотидов и расположена приблизительно в позициях -10 - -5 относительно старта трансляции, (в другой статье было указано, что старт SD обычно расположен в позиции -7 относительно старта инициации). В статье о полном геноме данной бактерии ШД не упоминается.
Поиск последовательности Шайн-Дальгарно был выполнен в бактерии Gordonibacte rpamelaeae 7-10-1-b .
Для осуществления поиска я нашла данную бактерию в БД Assembly на NCBI (ее последовательность), перешла на страницу последовательности в GeneBank и скачала там fasta файл с хромосомой ("send" => "Complete record", "File", "Fasta") и особенности (features), среди них есть CDSs ("send" => "Complete record", "File", "Feature Table"). После этого я преобразовала файл с Features в .xls формат с координатами кодирующих последовательностей, используя скрипт features2CDSs.py.
Затем было выбрано около 300 хороших генов (длина больше 300 п.н., аннотированные функции) для которых был создан fasta файл с областями поиска. Для создания позиционной матрицы весов (PWM) с помощью MEME я получила около 300 выбранных генов последовательности от -16 до 0 нуклеотида (от начала CDS = старта трансляции). Для поиска SD по всему геному я получила последовательности для всех CDS от -20 до 0 нуклеотида.
Далее я производила запуск сервиса MEME, где указала дополнительные параметры поиска: длина мотива от 6 до 10, 0 или 3 мотив на последовательность, искать мотив на той же цепи. Главным результатом работы сервиса является позиционно-весовая матрица (рисунок 1). HTML-выдача программы MEME - здесь.
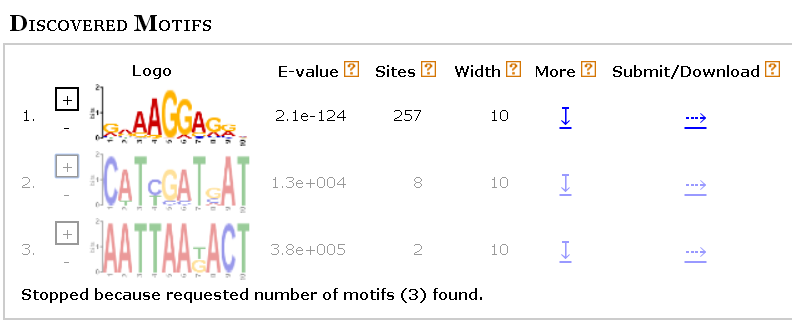
Рис. 1 Три разных мотива, найденных MEME.
Затем я произвела поиск в MEME с теми же параметрами, но уже только одного наилучшего мотива: meme_new.html. В результате была получена позиционная матрица весов (PWM).
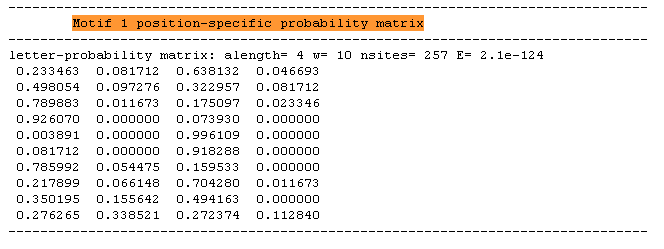
Рис. 2 Позиционная матрица весов для первого мотива.
![]()
Рис. 3 Лого найденных сигналов.
Затем уже проводился поиск по регионам перед всеми генами с помощью ресурса FIMO, предварительно расширив область поиска c -26 до -1 позиции до начала кодирующей последовательности. Найденная PWM была подана на вход алгоритму FIMO, который искал мотив последовательности Шайна-Дальгарно уже по всем генам. Порог E-value: 0.01. В результате было найдено 1510 генов, удовлетворяющих заданным условиям. Выдача программы приведена в таблице FIMOresults.xls.
Определение сайтов связывания транскрипционного фактора в участке хромосомы человекаКонтроль качества прочтений был сделан с помощью программы FastQC для полученных данных по ChIP-seq анализу. Результаты представлены по ссылке. График, отображающий качество нуклеотидов в чтениях, представлен на рисунке 6. Так как качество примерно одинаково по всей длине чтения и не сильно ухудшается к концу, а сами чтения достаточно короткие (36), обработка программой Trimmomatic не проводилась.
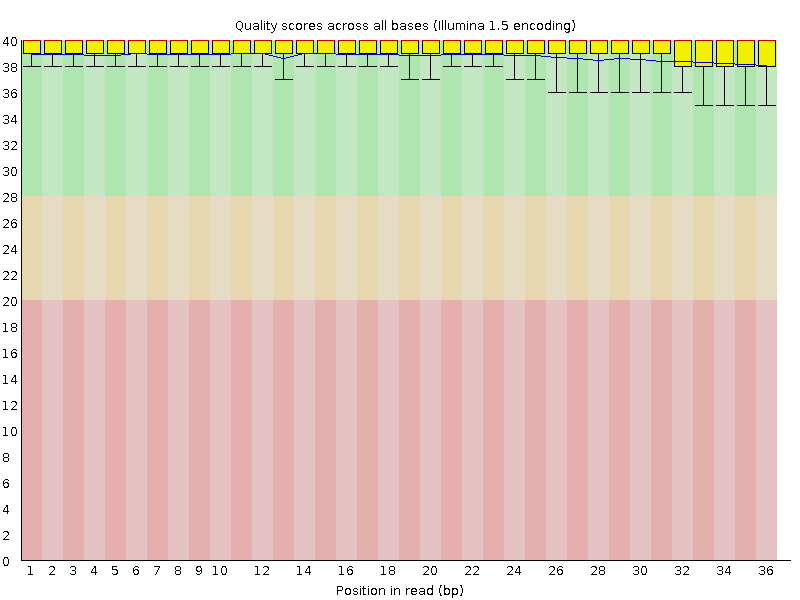
Рис. 6 Распределение качества нуклеотида в риде по положению в риде. Иизображение получено с помощью анализа качества чтения программой FastQC.
Затем чтения были откартированы на проиндексированный геном человека hg19 с помощью команды bwa mem ../hg19/GRCh37.p13.genome.fa chipseq_chunk25.fastq > chipseq_chunk25.sam. Далее были использованы следующие команды: samtools view -bSo chipseq_chunk25.bam chipseq_chunk25.sam (переводит выравнивание чтений с референсным геномов в бинарный формат, с которым потом работают программы), samtools sort chipseq_chunk25.bam -T chip_temp -o chipseq_chunk25.sorted.bam (сортирует выравнивание по координате начала чтения в референсе), samtools index chipseq_chunk25.sorted.bam (индексирует отсортированный файл), samtools idxstats chipseq_chunk25.sorted.bam > chipseq_chunk25.idxstats (записывает в файл chipseq_chunk25.idxstats информацию о количестве чтений, откартированных на каждый элемент генома) и samtools view -c chipseq_chunk25.sorted.bam (показывает, сколько чтений в сумме было откартированно на все элементы генома). Более подробно об использованных командах можно прочитать тут.
Всего было 7052 чтения, все они были откартированы на геном. При этом распределение чтений по хромосомам (рисунок 7) позволяет предположить, что я работаю с участками 14 хромосомы человека, так как больше всего чтений откартировано на 14 хромосому.
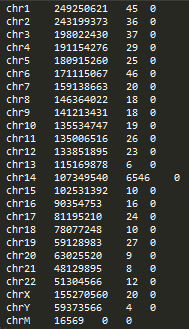
Рис. 7 Распределение числа откартированных чтений по хромосомам.
Затем я осуществила поиск пиков с помощью программы MACS. Так как пиков было слишком мало, я использовала команду macs2 callpeak -t chipseq_chunk25.sorted.bam -n chipseq_chunk25 --nomodel. В результате появились следющие файлы: chipseq_chunk25_peaks.narrowPeak, chipseq_chunk25_peaks.xls и chipseq_chunk25_summits.bed.
Всего найдено 5 пиков. Я визуализировала информацию из файла chipseq_chunk25_peaks.narrowPeak с помощью геномного браузера UCSC Genome Browser в разделе меню My Data > Custom Tracks. Для этого были добавлены следующие строки в файл: track type=narrowPeak visibility=3 db=hg19 name="my_peaks" description="Peaks from chunk 25" browser position chr14:69080000-69569000. Общее расположение пиков представлено на рисунке 8, а каждый пик в отдельности - на рисунках 9-13. Длина пика указана в подписи к соответствующему рисунку. Все пики обладают высокой достоверностью, судя по p-value и q-value. Расположение вершин пика относительно его крайних точек можно увидеть в 10 колонке файла chipseq_chunk25_peaks.narrowPeak (расстояние вершины от старта пика).
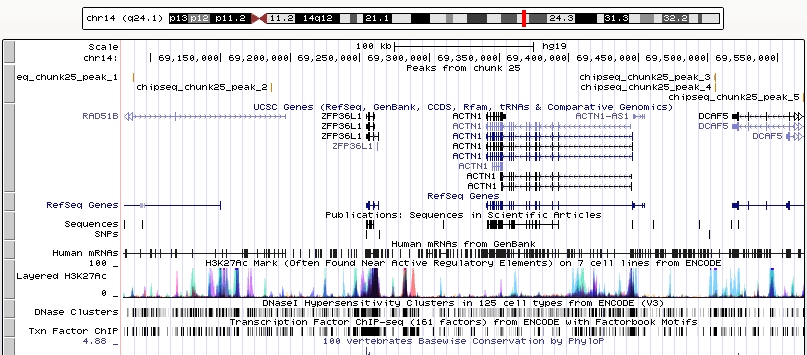
Рис. 8 Расположение найденных пиков в геноме человека (сборка hg19). Представлен участок 14 хромосомы. Изображение получено с помощью геномного браузера UCSC.
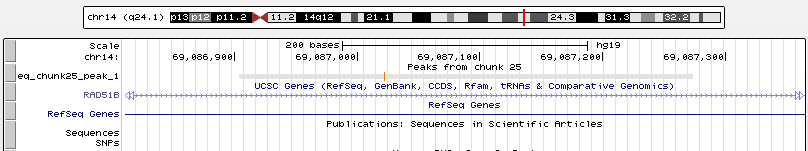
Рис. 9 Увеличенный первый пик, занимающий позиции chr14:69086905-69087274 и имеющий длину 370 nt. Первый пик попадает внутрь гена RAD51B.
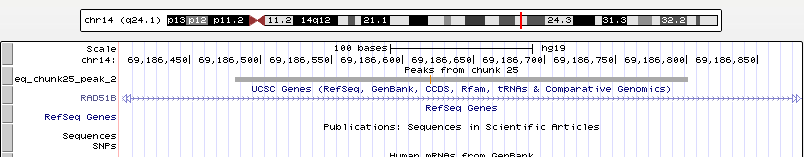
Рис. 10 Увеличенный второй пик, занимающий позиции chr14:69186483-69186801 и имеющий длину 319 nt. Второй пик попадает внутрь гена RAD51B.
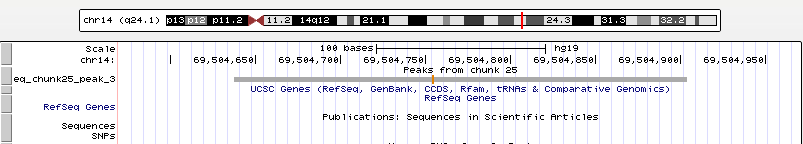
Рис. 11 Увеличенный третий пик, занимающий позиции chr14:69504638-69504904 и имеющий длину 267 nt. Третий пик попадает между генами ACTN1 и DCAF5.
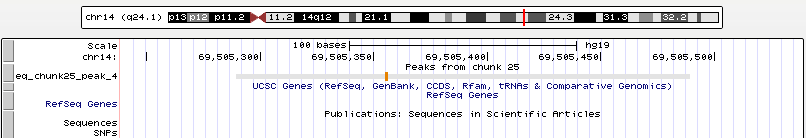
Рис. 12 Увеличенный четвертый пик, занимающий позиции chr14:69505290-69505489 и имеющий длину 200 nt. Четвертый пик попадает между генами ACTN1 и DCAF5.
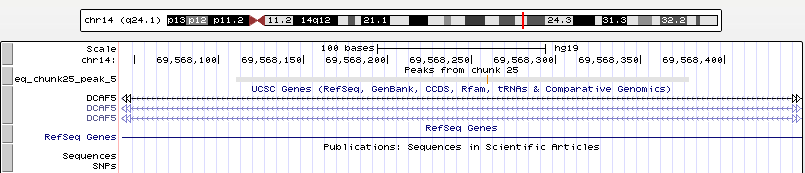
Рис. 13 Увеличенный пятый пик, занимающий позиции chr14:69568111-69568379 и имеющий длину 269 nt. Пятый пик попадает внутрь гена DCAF5.
Поиск сигналов TATA-бокс связывающего белка (TBP) в геноме человекаTATA-бокс связывающий фактор TBP - архейный и эукариотический белок, узнающий восьминуклеотидный сигнал в ДНК с консенсусом TATAWAAR. Он является одним из ключевых ДНК-узнающих белков при образовании на промоторе генов комплекса TFIID инициации транскрипции с помощью Pol II. Тем не менее, лишь часть промоторов имеет сигнал TATA-box, связываемый TBP.
С помощью геномного браузера UCSC я проанализировала результаты ChIP-seq анализа для TBP в стволовых эмбриональных клетках человека H1-hESC. Эксперимент проводился с использованием антител кролика (Рис. 14). TBP является субъединицей комплекса TFIID. TBP узнаетTATA-box и помогает направить инициаторный комплекс для РНК-полимераз II III.

Рис. 14 Описание эксперимента.
На рисунке 15 представлен ген, перед которым нет сигнала TATA-box (все сигналы в окрестности промотора гена примерно одинаковы и могут быть рассмотрены как шум). Это ген ALG1L2. Данный ген находится на 3 хромосоме (позиция chr3:129800674-129817233, длина гена 16560 п.о., он закодирован на (+)-цепи). На рисунке 15а показана вся промоторная область, на рисунке 15b - увеличенный участок, в котором должен был бы находиться сигнал TATA-box (с разрешением до отдельных нуклеотидов).
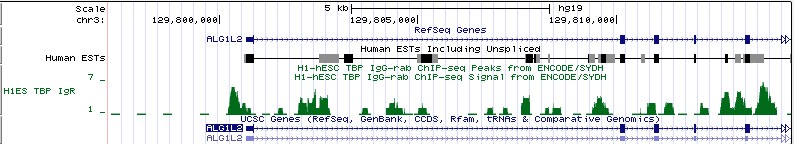
Рис. 15a Промоторная область гена ALG1L2.
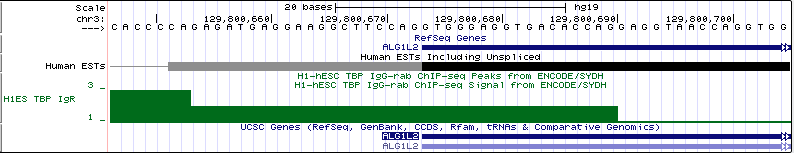
Рис. 15b Последовательность нуклеотидов в промоторной области гена ALG1L2.
На рисунке 16 представлен ген, перед которым есть сигнал TATA-box. Это ген KLF2. Данный ген находится на 19 хромосоме (позиция chr19:16435637-16439473, длина гена 3837 п.о., он закодирован на (+)-цепи). На рисунке 16а показана вся промоторная область, на рисунке 16b - увеличенный участок, в котором должен был бы находиться сигнал TATA-box (с разрешением до отдельных нуклеотидов).
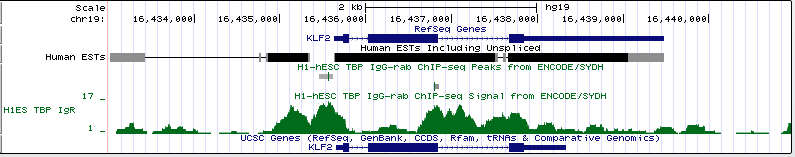
Рис. 16a Промоторная область гена KLF2.
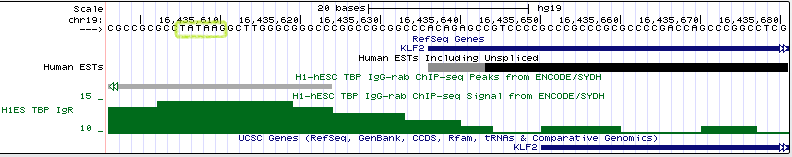
Рис. 16b Последовательность нуклеотидов в промоторной области гена KLF2. Зеленой рамкой обведен сайт связывания TBP.
На рисунке 17 представлен ген, перед которым есть сигнал TATA-box. Это ген H2AFX. Данный ген находится на 11 хромосоме (позиция chr11:118964585-118966177, длина гена 1593 п.о., он закодирован на (-)-цепи). На рисунке 17а показана вся промоторная область, на рисунке 17b - увеличенный участок, в котором должен был бы находиться сигнал TATA-box (с разрешением до отдельных нуклеотидов).
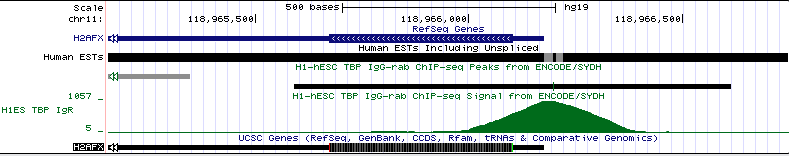
Рис. 17a Промоторная область гена H2AFX.
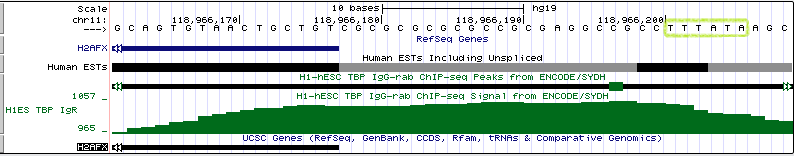
Рис. 17b Последовательность нуклеотидов в промоторной области гена H2AFX. Зеленой рамкой обведен сайт связывания TBP.
На рисунке 18 представлен ген, перед которым есть сигнал TATA-box. Это ген ITPR1. Данный ген находится на 3 хромосоме (позиция chr3:4535032-4889524, длина гена 354493 п.о., он закодирован на (+)-цепи). На рисунке 18а показана вся промоторная область, на рисунке 18b - увеличенный участок, в котором должен был бы находиться сигнал TATA-box (с разрешением до отдельных нуклеотидов).
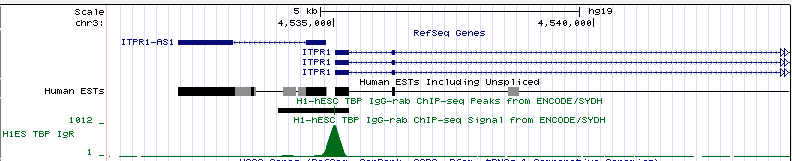
Рис. 18a Промоторная область гена ITPR1.
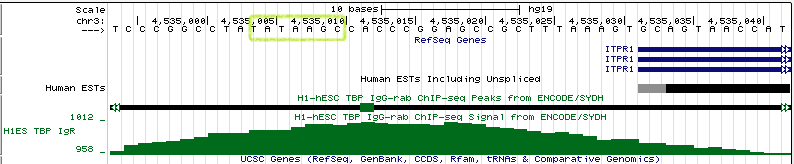
Рис. 18b Последовательность нуклеотидов в промоторной области гена ITPR1. Зеленой рамкой обведен сайт связывания TBP.
Во всех описанных случаях сигнал TATA-box расположен перед началом гена, поэтому по результатам данного анализа ChIP-seq TATA-box располагается перед геном.