1. Программа сравнения выравниваний
import sys
def read_file(filename):
seqs = []
current = ""
for line in open(filename):
line = line.strip()
if not line:
continue
if line[0] == '>':
if current:
seqs.append(current)
current = ""
else:
current = current + line
if current:
seqs.append(current)
return seqs
def compare(file1, file2, out=None):
s1 = read_file(file1)
s2 = read_file(file2)
if len(s1) != len(s2):
print(f"Ошибка: разное количество последовательностей!")
return
n = len(s1)
m1 = len(s1[0])
m2 = len(s2[0])
all_pairs = []
used_j = set()
for i in range(m1):
for j in range(m2):
if j in used_j:
continue
ok = True
for k in range(n):
if s1[k][i] != s2[k][j]:
ok = False
break
if ok:
all_pairs.append((i+1, j+1))
used_j.add(j)
break
all_pairs.sort()
blocks = []
single_pairs = []
if all_pairs:
block_start1 = all_pairs[0][0]
block_start2 = all_pairs[0][1]
prev1 = all_pairs[0][0]
prev2 = all_pairs[0][1]
for k in range(1, len(all_pairs)):
curr1, curr2 = all_pairs[k]
if curr1 == prev1 + 1 and curr2 == prev2 + 1:
prev1 = curr1
prev2 = curr2
else:
block_len = prev1 - block_start1 + 1
if block_len >= 2:
blocks.append((block_start1, prev1, block_start2, prev2))
else:
single_pairs.append((block_start1, block_start2))
block_start1 = curr1
block_start2 = curr2
prev1 = curr1
prev2 = curr2
block_len = prev1 - block_start1 + 1
if block_len >= 2:
blocks.append((block_start1, prev1, block_start2, prev2))
else:
single_pairs.append((block_start1, block_start2))
if out:
f = open(out, 'w')
f.write("Блоки одинаково выровненных колонок:\n")
for b in blocks:
f.write(f"({b[0]},{b[1]})=({b[2]},{b[3]})\n")
if single_pairs:
f.write("\nОдиночные одинаковые колонки:\n")
for p in single_pairs:
f.write(f"{p[0]}\t{p[1]}\n")
f.close()
print(f"Результат записан в: {out}")
total_in_blocks = sum(b[1] - b[0] + 1 for b in blocks)
print(f"Длина 1 выравнивания: {m1}")
print(f"Длина 2 выравнивания: {m2}")
print(f"Найдено блоков: {len(blocks)}")
print(f"Колонок в блоках: {total_in_blocks}")
print(f"Одиночных колонок: {len(single_pairs)}")
print(f"Всего одинаковых колонок: {total_in_blocks + len(single_pairs)}")
print(f"% в блоках от 1 выравнивания: {round(100*total_in_blocks/m1, 1)}")
print(f"% в блоках от 2 выравнивания: {round(100*total_in_blocks/m2, 1)}")
if len(sys.argv) == 2 and sys.argv[1] == '-h':
print('Использование: python compare.py файл1 файл2 [выход]')
print('Файлы: выравнивание в формате FASTA')
print('Выход: блоки (s1,f1)=(s2,f2) длиной >= 2')
print(' и список одиночных одинаковых колонок')
elif len(sys.argv) >= 3:
out = sys.argv[3] if len(sys.argv) > 3 else None
compare(sys.argv[1], sys.argv[2], out)
else:
print('Укажите два файла. -h для справки')
2. Сравнение трёх программ MSA на примере фосфолипаз A2
Белки:
P00602 — Naja mossambica (кобра)
Q3C2C1 — Acanthaster planci (терновый венец)
P00630 — Apis mellifera (пчела)
Длины выравниваний:
ClustalO: 212 колонок, MAFFT: 228 колонок, MUSCLE: 193 колонки.
Результаты попарного сравнения (программа compare.py)
python compare.py mafft.fa muscle.fa ma_mu.txt
Результат записан в: ma_mu.txt
Длина 1 выравнивания: 228
Длина 2 выравнивания: 193
Найдено блоков: 7
Колонок в блоках: 65
Одиночных колонок: 36
Всего одинаковых колонок: 101
% в блоках от 1 выравнивания: 28.5
% в блоках от 2 выравнивания: 33.7
Блоки одинаково выровненных колонок:
(1,8)=(1,8)
(17,23)=(17,23)
(38,64)=(38,64)
(66,78)=(66,78)
(82,84)=(82,84)
(88,92)=(88,92)
(164,165)=(172,173)
Одиночные одинаковые колонки:
15 15
16 10
28 28
35 13
36 36
65 34
79 118
80 165
81 117
86 93
94 24
95 95
97 86
100 119
102 103
105 11
116 96
126 131
128 29
python compare.py mafft.fa clustalo.fa ma_cl.txt
Результат записан в: ma_cl.txt
Длина 1 выравнивания: 228
Длина 2 выравнивания: 212
Найдено блоков: 8
Колонок в блоках: 61
Одиночных колонок: 68
Всего одинаковых колонок: 129
% в блоках от 1 выравнивания: 26.8
% в блоках от 2 выравнивания: 28.8
Блоки одинаково выровненных колонок:
(29,34)=(33,38)
(53,55)=(67,69)
(64,75)=(54,65)
(85,95)=(75,85)
(123,125)=(108,110)
(155,164)=(139,148)
(170,173)=(154,157)
(179,190)=(163,174)
Одиночные одинаковые колонки:
3 27
6 26
8 18
13 11
18 112
19 103
21 121
22 117
23 111
28 149
36 102
51 70
52 197
56 177
57 185
58 71
61 159
62 66
Muscle
Mafft
ClustalO
| Сравнение | Длина 1 | Длина 2 | Блоков | В блоках | Одиночных | Всего | % в блоках (1) | % в блоках (2) |
|---|---|---|---|---|---|---|---|---|
| MAFFT vs MUSCLE | 228 | 193 | 7 | 65 | 36 | 101 | 28.5% | 33.7% |
| MAFFT vs ClustalO | 228 | 212 | 8 | 61 | 68 | 129 | 26.8% | 28.8% |
Анализ
- MAFFT делает самые длинные выравнивания (228 колонок) — вставляет больше гэпов.
- MUSCLE делает самые компактные выравнивания (193 колонки) — меньше гэпов.
- ClustalO занимает промежуточное положение (212 колонок).
- MAFFT vs ClustalO: 129 одинаковых колонок (больше всего), 8 блоков, 61 колонка в блоках (26.8–28.8%).
- MAFFT vs MUSCLE: 101 одинаковая колонка, 7 блоков, 65 колонок в блоках (28.5–33.7%). Выше процент в блоках относительно MUSCLE (33.7%).
- MAFFT (B) больше похож на ClustalO (A): 129 общих колонок против 101 у MUSCLE. Ближе по длине выравнивания (228 vs 212, разница 16 колонок; у MUSCLE 193 — разница 19).
3. Сравнение структурного (PDBeFold) и последовательностного (MUSCLE) выравнивания для домена Zn_clus
Выбраны 3 белка (PDB ID): 1PYI, 1AW6, 2ERG
python compare.py msa.fa pdbefold.fasta msa_pdbefold.txt
Результат записан в: msa_pdbefold.txt
Длина 1 выравнивания: 162
Длина 2 выравнивания: 162
Найдено блоков: 4
Колонок в блоках: 8
Одиночных колонок: 7
Всего одинаковых колонок: 15
% в блоках от 1 выравнивания: 4.9
% в блоках от 2 выравнивания: 4.9
Блоки одинаково выровненных колонок:
(93,94)=(92,93)
(97,98)=(96,97)
(103,104)=(102,103)
(116,117)=(115,116)
Одиночные одинаковые колонки:
101 100
112 122
114 113
119 118
121 120
123 111
124 123
PDBeFold
Muscle
Проект в Jalview
Результат сравнения двух выравниваний — структурного (PDBeFold) и по последовательности (MUSCLE) — для трёх белков домена Zn_clus показывает очень низкое перекрытие: всего 15 колонки выровнены одинаково (из 162 позиций), что составляет 4.9% от длины выравнивания. Оба выравнивания были покрашены Clustal и By Conservation (100%)
Рисунок 1. Структурное выравнивание в Jalview
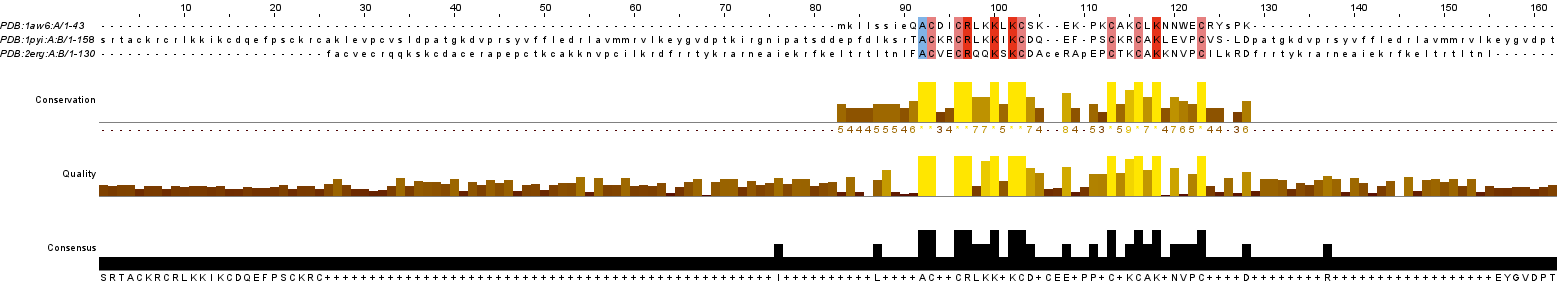
Рисунок 2. Выравнивание MUSCLE в Jalview
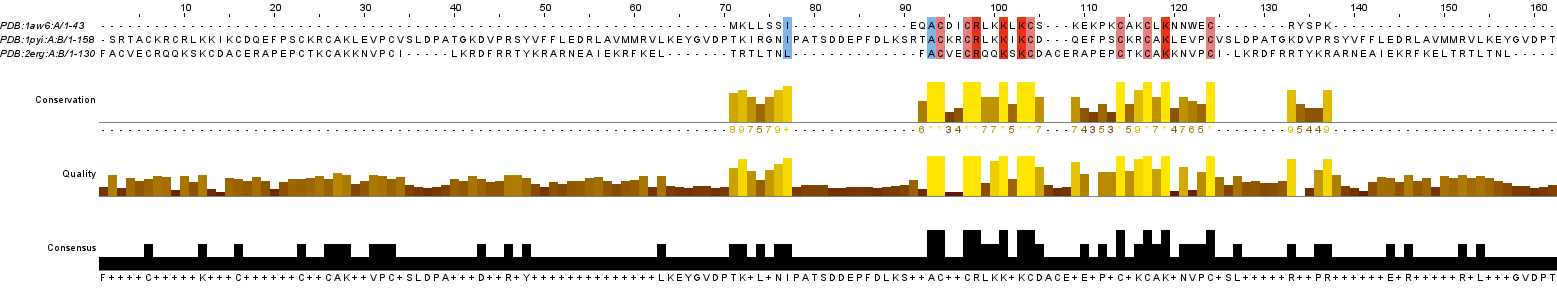
При сопоставлении выравниваний найдено 2 длинных не абсолютно консервативных блока (оба принадлежат цистеиновому мотиву):
1. 92-103 (для PdBeFold), 93-104 (для MUSCLE) - содержит 7 абсолютно консервативнх колонок с такими консенсусным аминокислотами, как: цистеин (3), лизин (2), аланин (1), аргинин (1)
2. 113-123 (для PdBeFold), 114-124 (для MUSCLE) - содержит 4 абсолютно консервативные колонки с такими консенсусными аминокислотами, как: цистеин (3), лизин (1)
Отличие: выравнивание программой MUSCLE выделило колонку с номером 77 (полуконсервативная, в последовательностях белков 1AW6 и 1PYI содержит изолейцин, а в 2ERG - лейцин)
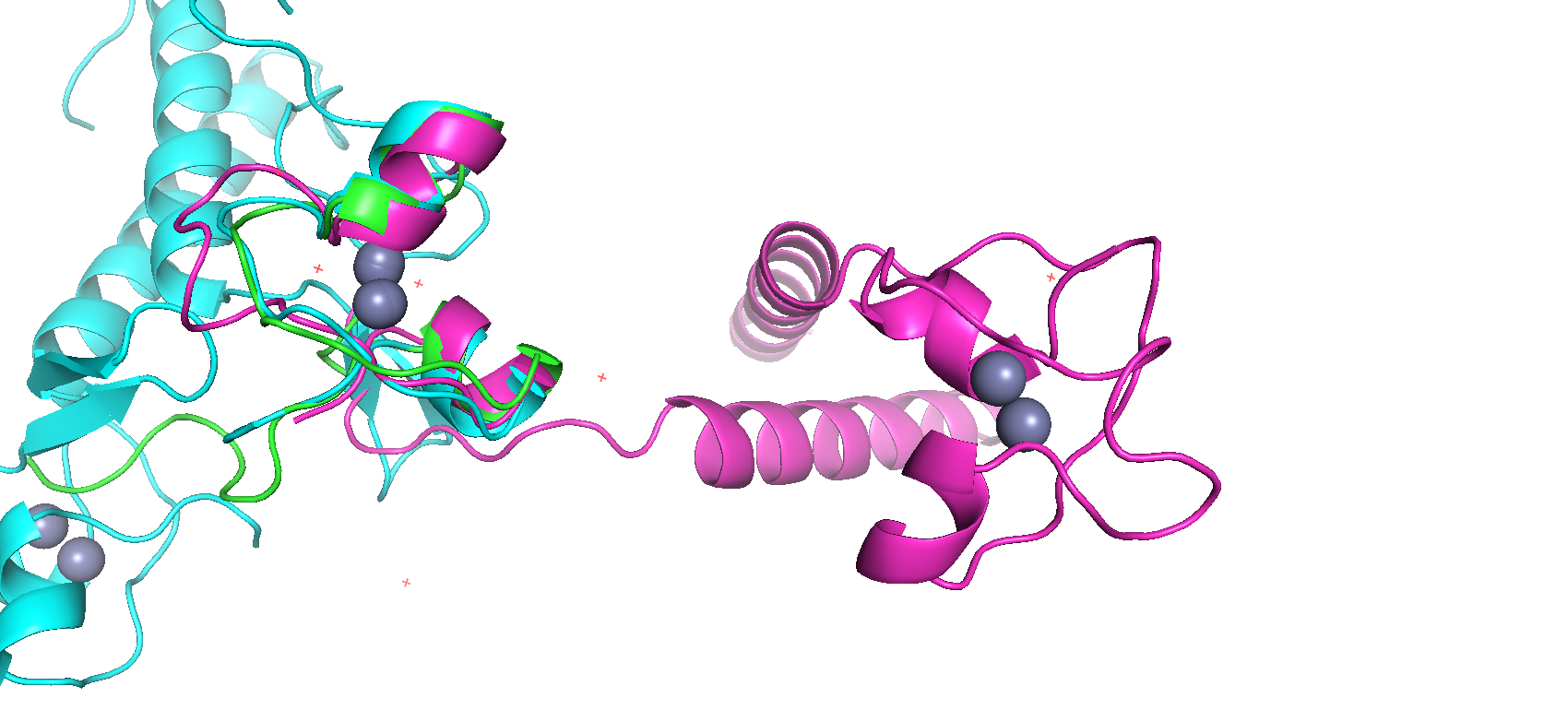
Исходя из структурного выравнивания, можно сделать вывод, что перекрывающиеся по цветам области белка составляют часть цинкового кластера (на рисунке рядом с альфа-спиралями расположены 2 атома цинка), который и образует Zn_clus, то есть выбранные белки гомологичны по этому домену (из постановки задачи)
Рисунок 3. Пространственное сравнение структур в Pymol
4. Краткое описание программы MSA: MUSCLE
MUSCLE (сокращение от MUltiple Sequence Comparison by Log‑Expectation) — популярный инструмент для множественного выравнивания нуклеотидных и белковых последовательностей. Алгоритм построения выравнивания выглядит следующим образом: сначала MUSCLE делает черновое выравнивание, оценивая расстояния между последовательностями через k‑меры и применяя прогрессивный метод. А потом начинает улучшать результат итеративно — пересчитывает профили, удаляет последовательности, переставляет их, чтобы добиться более точного выравнивания. В основе всего этого лежит log‑expectation score (логарифмическое ожидание), который как раз и повышает точность.
Рисунок 4. Алгоритм построения выравнивания MUSCLE

Отличие от других программ MSA:
Отчёт по практикуму 12 | Мисюрёва Анастасия | 2026