Сборка de novo
Мне был предоставлен код доступа проекта по секвенированию бактерии Buchnera aphidicola SRR4240361. Я получила чтения, полученные в проекте, по ссылке для скачивания со страницы https://www.ebi.ac.uk/ena/browser/view/SRR4240361. Это короткие (длины 39) чтения, полученные по технологии Illumina. Далее была создана специальная дирректория для работы: /mnt/scratch/NGS/nsv2002.
Далее был скачен архив с чтениями в рабочую директорию, это было сделано с помощью команды wget
Команда выглядела так: wget ftp://ftp.sra.ebi.ac.uk/vol1/fastq/SRR424/001/SRR4240361/SRR4240361.fastq.gz
1. Подготовка чтений программой trimmomatic.
Прежде всего надо было удалить возможные остатки адаптеров, адаптеры для Illumina собраны в файлах в директории /mnt/scratch/NGS/adapters. Для удобства эти файлы были скачены в рабочую дирректорию с помощью команды cat. Команда выглядела так: cat /mnt/scratch/NGS/adapters/* >> adapters.fa
Затем с помощью программы trimmomatic были удалены возможные остатки адаптеров командой: java -jar /usr/share/java/trimmomatic.jar SE SRR4240361.fastq.gz cut.fastq.gz -trimlog trim.log ILLUMINACLIP:adapters.fa:2:7:7
Полученные данные: Input Reads: 7272621 Surviving: 7238089 (99.53%) Dropped: 34532 (0.47%)
После этого были удалены с правых концов чтений нуклеотиды с качеством ниже 20 и были оставлены только такие чтения, длина которых не меньше 32 нуклеотидов с помощью команды: java -jar /usr/share/java/trimmomatic.jar SE trimmed.fastq.gz cut2.fastq.gz -trimlog trim2.log TRAILING:20 MINLEN:32.
Полученные данные: Input Reads: 7238089 Surviving: 6834335 (94.42%) Dropped: 403754 (5.58%)
Из полученных данных можно сделать следующие выводы:
1)Так как в первый раз отфильтровалось только 0,47%, то это может означать, что адаптеров в чтениях практически не было.
2)Предоставленные данные обладают хорошим качеством, так как была удалена достаточно малая часть.
Изначальный файл весил - 193M. Вес файла после выполнения первой команды - 192M (совсем чуть-чуть уменьшился). После выполнения второй команды размер файла также незначительно уменьшился - 178M. Можно сделать вывод, что изначально файлы уже были высокого качества(были уже очищенными от адаптеров), поэтому после выполнения команд их вес уменьшился незначительно. Вес файлов можно узнать c помощью команды: ls -lh
2. Работа с командой velvet.
Далее было необходимо разобраться с работой программ velveth и velvetg. Velveth на основе заданного файла должна подготовить k-меры длины k=31 (максимально возможной при нашей длине чтений). Необходимо было учитывать, что в нашем случае чтения короткие и не парные. Cначала была созадана папка kmers, где хранятся К-меры длины 31. Это было сделано с помощью команды: velveth kmers 31 -fastq.gz cut2.fastq.gz -short.
Затем была произведена сборка по К-мерам, которые были получены на предыдущем шаге, записываем информацию в файл: assembly.log. Команда: velvetg kmers &> assembly.log
Результаты: assembly.log
Из этого файла можно узнать значение параметра N50 - 25683.
velvetg также создаёт в ранее упомянутой папке kmers файлы с информацией о полученных контигах stats.txt и файл с их последовательностями contigs.fa.
Из этих файлов были выбраны 3 контига наибольшей длины:
Таблица 1. 3 самых больших контига.
| ID | Длина | Покрытие | fasta файл |
|---|---|---|---|
| 6 | 49238 | 26,66 | contig 6 |
| 2 | 45555 | 26,45 | contig 2 |
| 34 | 43866 | 23,52 | contig 34 |
При анализе файла stats.txt были также выявлены контиги с аномально малым покрытием(более чем в 5 раз отличающимся от "типичного"). Три из них:
Таблица 2. 3 контига с аномально низким покрытием.
| ID | Длина | Покрытие |
|---|---|---|
| 462 | 2 | 1,00 |
| 469 | 3 | 1,33 |
| 477 | 4 | 1,50 |
3. Анализ.
Далее было произведено сравнение программой megablast каждого из трёх самых длинных контигов с хромосомой Buchnera aphidicola (GenBank/EMBL AC — CP009253).
С полученными результатами можно ознакомиться ниже:
Таблица 3. Информация о выравниваниях.
| ID контига | E-value | Query Cover | Identities | Total Score |
|---|---|---|---|---|
| 6 | 0.0 | 80% | 75% | 21380 |
| 2 | 0.0 | 70% | 77% | 17272 |
| 34 | 0.0 | 71% | 79% | 16707 |
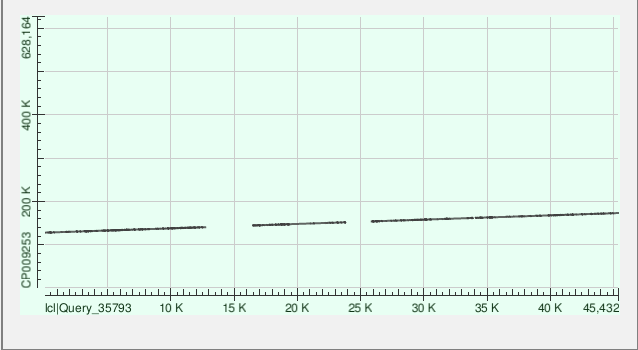
Контиг 6 соответствует референсному геному на участке с 127825 по 140555 нуклеотид. В лучшем выравнивании - 548 гэпов (4%).
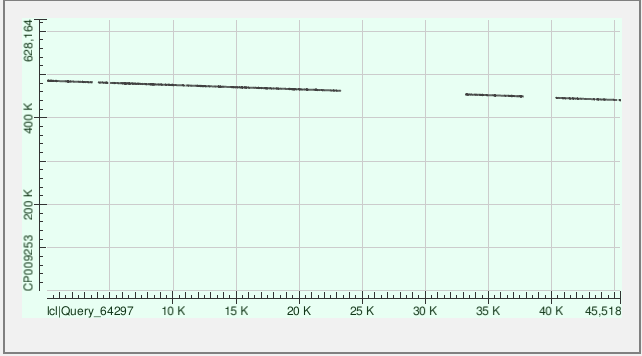
Контиг 2 соответствует референсному геному на участке с 467412 по 474667 нуклеотид. В лучшем выравнивании - 208 гэпов (2%).
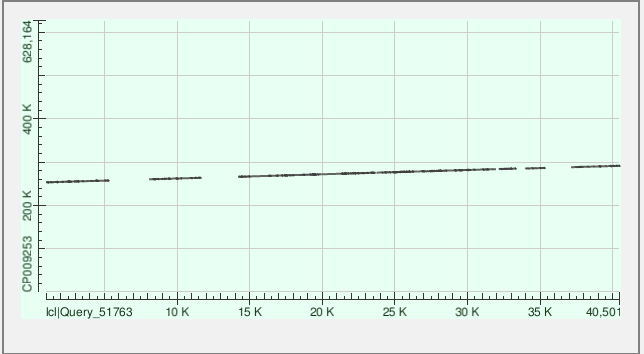
Контиг 34 соответствует референсному геному на участке с 266073 по 275551 нуклеотид. В лучшем выравнивании - 361 гэпов (3%).