
Парные выравнивания белков. Применение алгоритмов парных выравниваний к белку YQGN_BACSU
Сравнение матриц аминокислотных замен
Матрицы типа BLOSUM построены с использованием разработанной авторами (Steven Henikoff & Jorja Henikoff) ранее базы данных BLOCKS, в которой содержатся блоки выравниваний цитоплазматических белков из нескольких сотен семейств. В блоках могут встречаться практически идентичные последовательности, за счет которых частоты пар могут быть ошибочно «сдвинуты», поэтому их кластеризуют (объединяют). Матрицу строят на основе последовательностей, процент идентичности которых меньше заданного заранее значения, указанного в конце названия матрицы. Например, BLOSUM62.Матрица типа PHAT (predicted hydrophobic and transmembrane regions) - a Transmembrane-Specific Substitution Matrix - матрица для трансмембранных участков белков. Выделение отдельно этой матрицы нужно потому, что у трансмембранных участков белков, по сравнению с цитоплазматическими, сильно изменены частоты аминокислот.
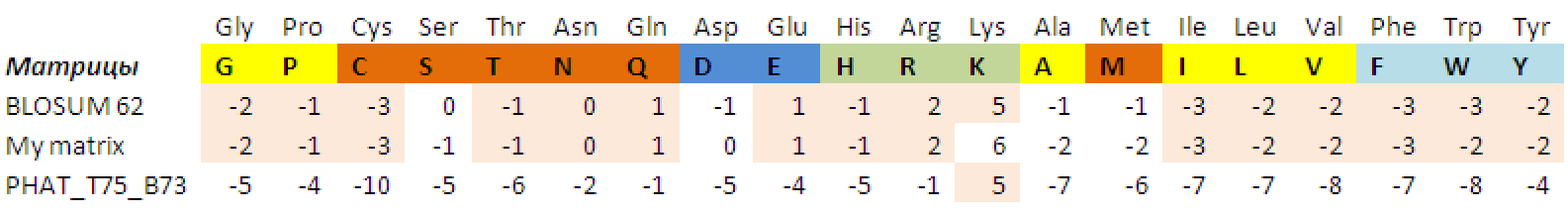
| Табл. 1. Сравнение матриц аминокислотных замен для лизина (Lys, L) |
|
 |
 |
(увеличенное изображение при клике)
В таблице представлен вес замен лизина на самого себя и остальные протеиногенные аминокислоты для трех разных матриц. Аминокислоты выделены по их функциональной группе. Среди веса замен выделены идентичные значения.
Данные по весам замен матрицы BLOSUM62 и полученной в ходе работы матрицы отличаются всего по нескольким аминокислотам: Ser, Asp, Ala, Met, Lys. Это можно объснить тем, что матрица BLOSUM62 использовала более старую базу данных последовательностей, чем использовалась для построения новой матрицы. Поэтому появились новые выравненные последовательности, которые могли повлиять на количество сопоставлений для всех пар аминокислот в блоках.
Сравнение выравниваний, полученных для коротких мутантов вручную и построенных классическими алгоритмами Нидлмана-Вунша и Смита-Ватермана
В программе needle из пакета EMBOSS реализован алгоритм Нидлмана-Вунша[2], а в программе water - Смита-Ватермана[2]. Эти алгоритмы предназначены для выравнивания нескольких последовательностей. Программа needle позволяет посторить глобальное выравнивание, а программа water - локальное. В чем же выражается их отличие?Ниже представлены параметры, которые использует каждый алгоритм, стандартные значения этих параметров приведены в [].
water:
-gapopen [10.0] (Number from 0.000 to 100.000) - штраф за открытие гэпа.
-gapextend [0.5 - штраф за последующие гэпы после открытия.
-datafile [EBLOSUM62 для белков, EDNAFULL для ДНК] - матрица весов замен
needle:
-endextend [0.5] (Floating point number from 0.0 to 10.0) - штраф за продолжение концевого гэпа
-endopen [10.0] - штраф за начало концевого гэпа
-endweight - штрафы за кончевые гэпы (применять/не применять) [применять]
-gapopen [10.0] (Number from 0.000 to 100.000) - штраф за открытие гэпа.
-gapextend [0.5] - штраф за последующие гэпы после открытия.
-datafile [EBLOSUM62 для белков, EDNAFULL для ДНК] - матрица весов замен.
Теперь сравним выравнивания, полученные для коротких мутантов вручную (см. файл) и построенные классическими алгоритмами Нидлмана-Вунша и Смита-Ватермана. Результаты для каждого из мутантов приведены на рис. 1-3.

рис.1 Мутант № 1_1
Вероятность изменения остатка - 0,6.
Вероятность замены остатка в случае, если данная позиция будет изменена - 0,6.
(Увеличенное изображение при клике на картинке)
Координаты участка выравнивания для полной последовательности YQGN_BACSU для needle - 137 - 158.
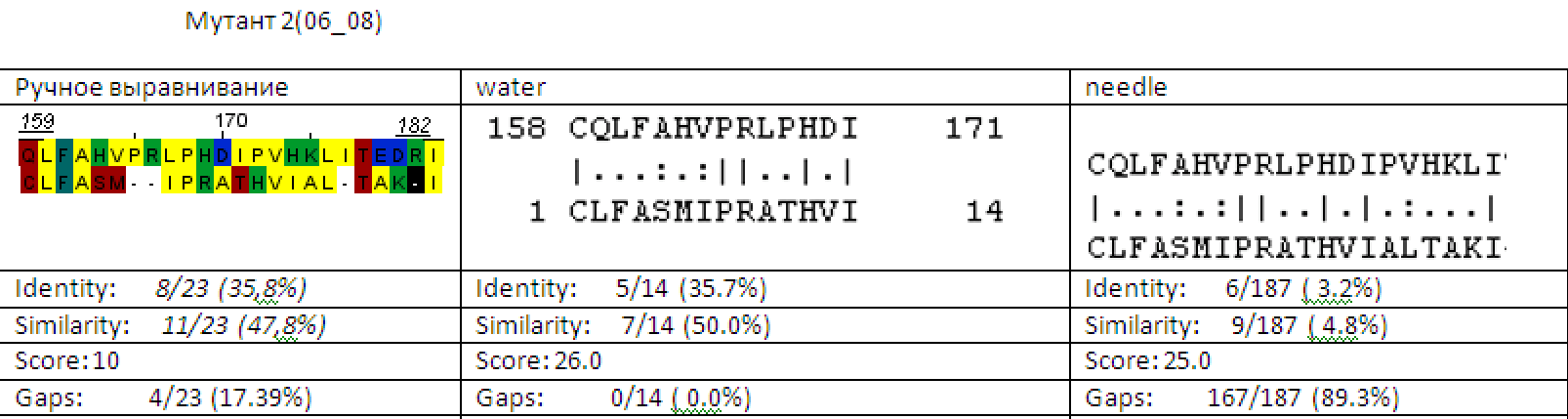
рис.2 Мутант № 2_1
Вероятность изменения остатка - 0,6.
Вероятность замены остатка в случае, если данная позиция будет изменена - 0,8.
(Увеличенное изображение при клике на картинке)
Координаты участка выравнивания для полной последовательности YQGN_BACSU для needle - 158 - 177.

рис.3 Мутант № 3_1
Вероятность изменения остатка - 0,4.
Вероятность замены остатка в случае, если данная позиция будет изменена - 0,8.
(Увеличенное изображение при клике на картинке)
Координаты участка выравнивания для полной последовательности YQGN_BACSU для needle - 118 - 137.
Приложения:
Список литературы:
- Needleman SB, Wunsch CD. A general method applicable to the search for similarities in the amino acid sequence of two proteins. J Mol Biol. 1970 Mar;48(3):443-53. PMID: 5420325. Версия PDF
- Алгоритм Нидлмана-Вунша, написанный на языке Ruby
- Описание программ needle, water пакета EMBOSS.
© Nuzhdina Ekaterina, 2012