
Комплексы ДНК-белок
Поиск ДНК-белковых контактов в структуре 1DDN
В JMol с помощью команды define
можно задавать множества атомов. Определим следующие множества:
- множество атомов кислорода 2'-дезоксирибозы (set1).
- множество атомов кислорода в остатке фосфорной кислоты (set2).
- множество атомов азота в азотистых основаниях (set3).
- всей структуры
- только ДНК в проволочной модели
- проволочная модель ДНК с выделенными шариками множества атомов set1
- проволочная модель ДНК с выделенными шариками множества атомов set2
- проволочная модель ДНК с выделенными шариками множества атомов set3
Ссылка на script.
Описанние ДНК-белковых контактов в структуре 1DDN
Опишем ДНК-белковые контакты в заданной структуре. Сравним количество контактов разной природы.Будем считать полярными атомы кислорода и азота, а неполярными атомы углерода, фосфора и серы.
Назовем полярным контактом ситуацию, в которой расстояние между полярным атомом белка и полярным атомом ДНК меньше 3.5 Å, а неполярным контактом будем считать пару неполярных атомов на расстоянии меньше 4.5 Å.
Определим число контактов и заполним таблицу 1.
Таблица 1. Контакты разного типа в комплексе 1DDN.pdb
Контакты атомов белка с |
Полярные |
Неполярные |
Всего |
остатками 2'-дезоксирибозы |
7 |
31 |
38 |
остатками фосфорной кислоты |
46 |
26 |
72 |
остатками азотистых оснований со стороны большой бороздки |
7 |
1 |
8 |
остатками азотистых оснований со стороны малой бороздки |
0 |
1 |
1 |
Получение схемы ДНК-белковых контактов с помощью программы nucplot
Программа nucplot, предназначенная для визуализации контактов между ДНК и белком, запускается на сервере kodomo. К сожалению, программа работает только со старым форматом PDB, поэтому используем программу remediator для перевода.
nucplot 1DDN_old.pdb
В результате выполенения программы будут созданы файлы:
- 1DDN_old.hb2 - H-bonds
- 1DDN_old.nb2 - non-bonded contacts
- 1DDN_old.bond - Bonds file
- nucplot.ps
- nucplot.par
- hb2.log
- hbadd.log
- nb2.log
Также программа выводит количество цепей ДНК(2) и белка(8) в структуре, при запуске в самом терминале.
Но нас интересует файл nucplot.ps (ps = формат Postscript).
Откроем его ассоциированной программой
GSview. Мы получим
изображение популярной схемы ДНК-белковых контактов.
P.S. Удобнее открывать формат ps оказалось онлайн-вьювером.
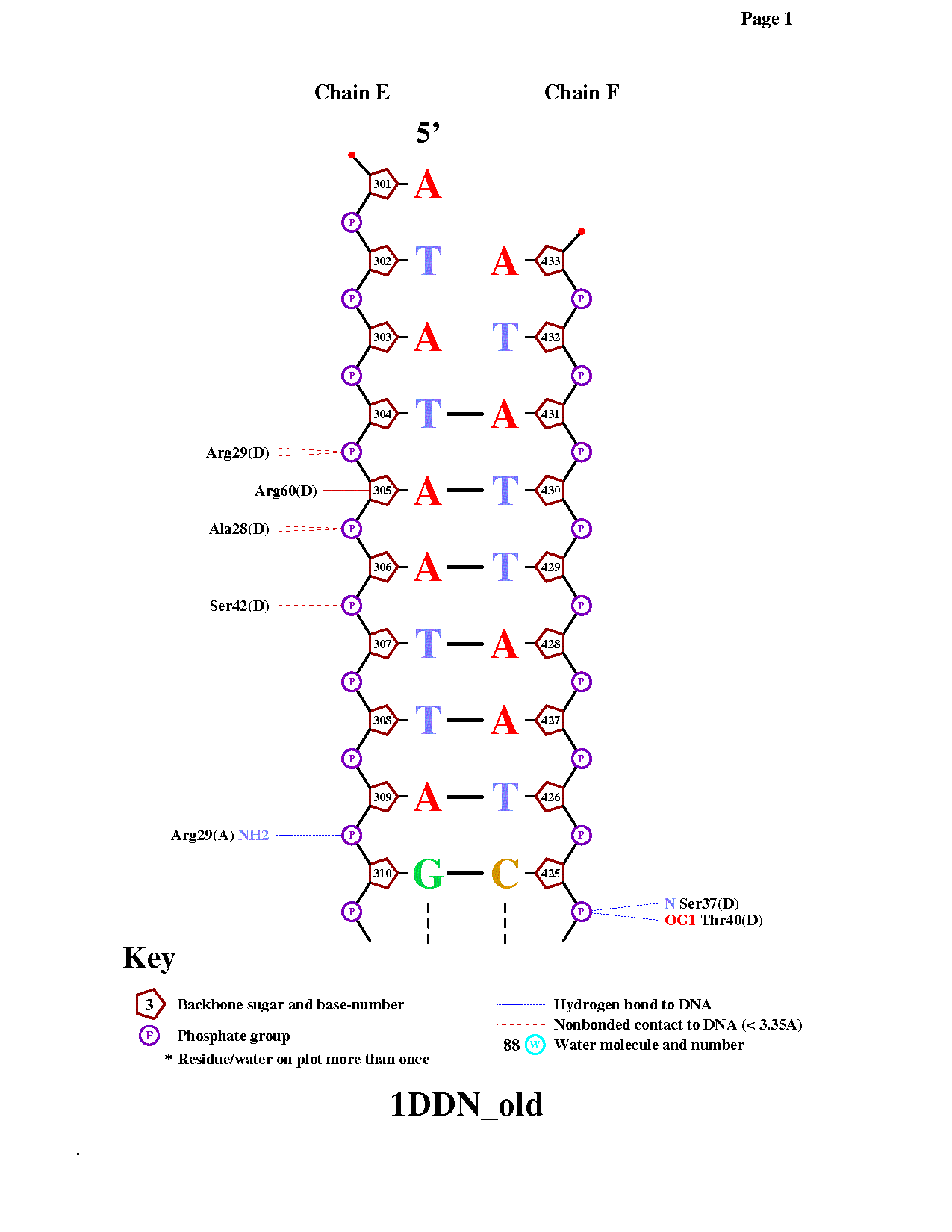
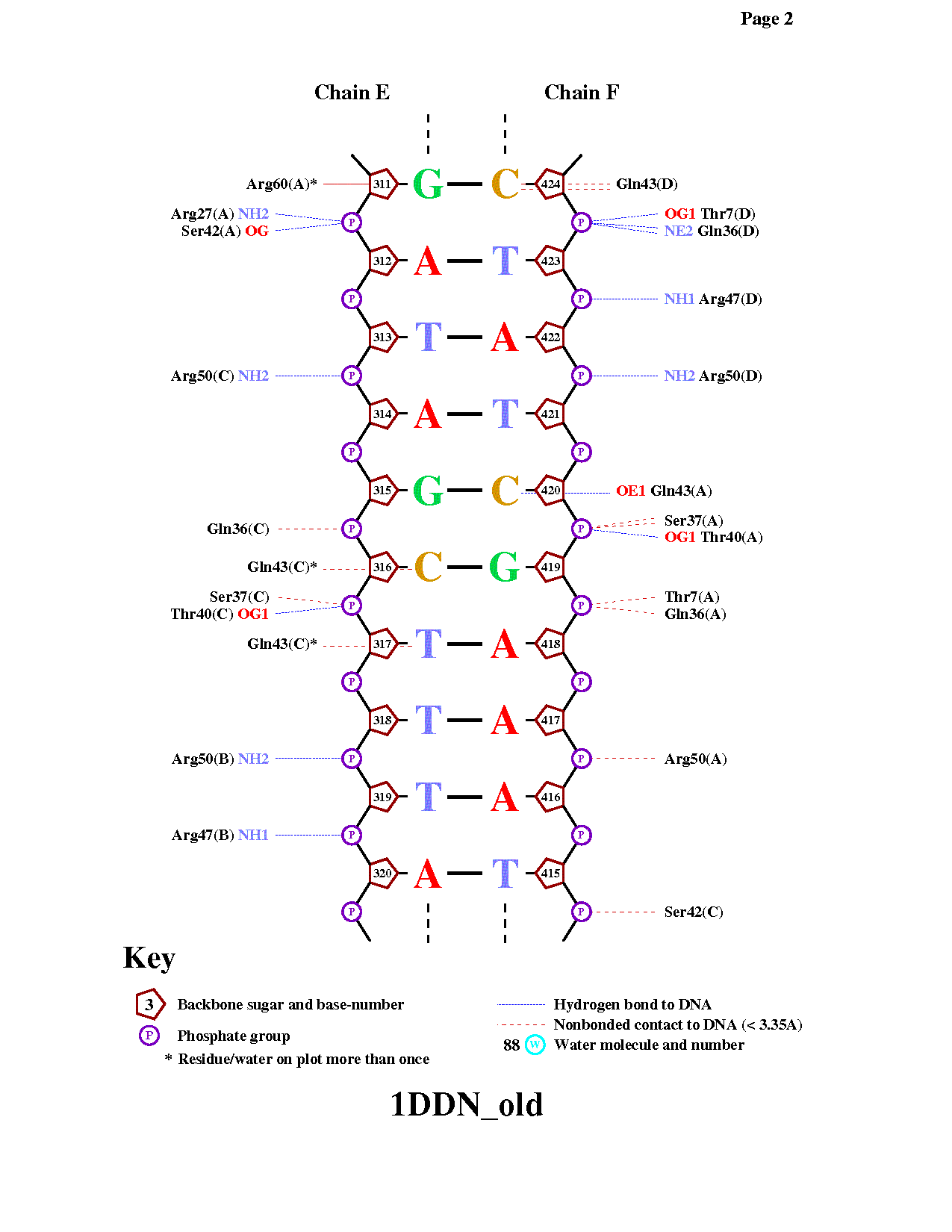
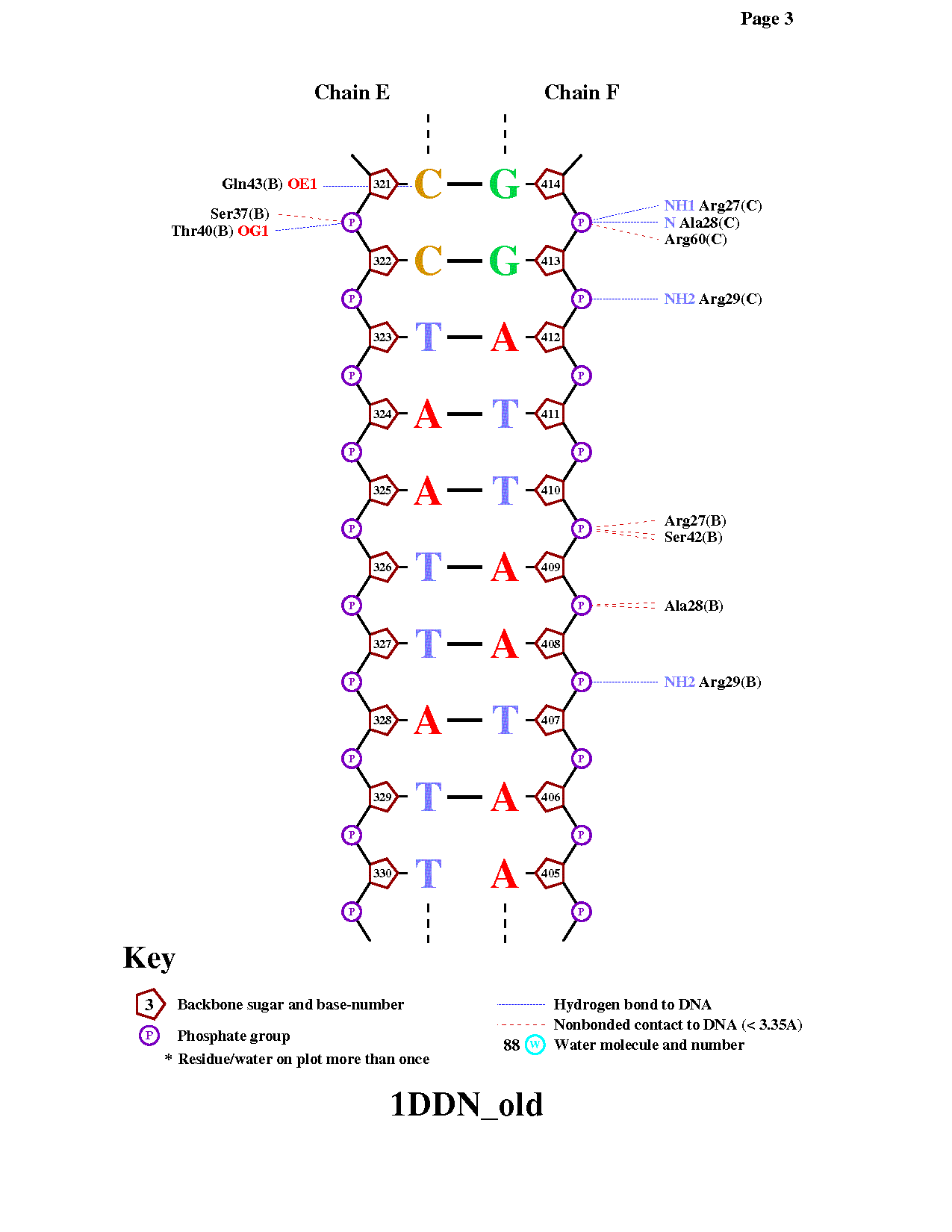
Рис.1 Популярная схема ДНК-белковых контактов, полученная с помощью программы nucplot
 |
 |
 |
 |
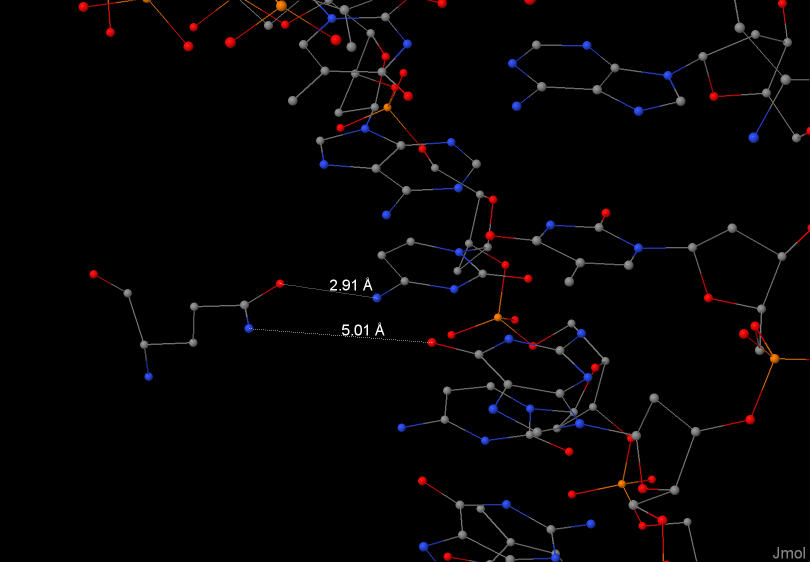
Из рис.1 видно, что аминокислотный остаток с наибольшим числом указанных на схеме контактов с ДНК - Arg29 (3 контакта)
Аминокислотный остаток наиболее важный для распознавания последовательности ДНК должен взаимодействовать
с азотистым основанием. Gln43:С образует целых два таких взаимодействия.
Предсказание вторичной структуры 1G59 тРНК
Предсказание вторичной структуры тРНК путем поиска инвертированных повторов
Программа einverted из пакета EMBOSS позволяет найти инвертированные участки в нуклеотидных последовательностях.
На вход программе дается файл с последовательностью тРНК в fasta-формате.
Найдем возможные комплементарные участки в последовательности структуры 1G59 тРНК.
Сравним результаты с данными, полученными
ранее с помощью find_pair
(см. 3 семестр -> A- и В- формы ДНК. Структура РНК).
Результаты сравнения занесем в таблицу 2, приведенную ниже.
einverted -sequence 1G59.fasta
Вывод программы в терминале:
Finds inverted repeats in nucleotide sequences
Gap penalty [12]: *
Minimum score threshold [50]: *
Match score [3]: *
Mismatch score [-4]: *
Sanger Centre program inverted output file [sequence.inv]:
File for sequence of regions of inverted repeats. [sequence.fasta]:
Примечание: * - это вводимый пользвателем параметр. Если не вводить свои значения, а просто нажимать Enter
то программа идет со стандартными настройками.
При стандартных установках программа не находила комплементарных пар, но задав Minimum score threshold = 15,
был получен файл seq.inv. Из файла видно, что найден только акцепторный стебель,
причем не весь и немного смещенный, по сравнению с результатами, найденнными программой find pair.
Найдены спаренные основания 3-7, 65-69, а программой find pair найденны 1-7, 66-72.
Предсказание вторичной структуры тРНК по алгоритму Zucker
Программа mfold реализует алгоритм Зукера. Воспользуемся web вариантом
ссылка.
Параметр P - единственный, который имеет смысл менять. Он указывает, на сколько процентов выдаваемое предсказание структуры
может отличаться по своей вычисленной энергии от оптимального.
Чем больше значение этого параметра, тем больше вариантов предсказания будет выдано.
Были получены результаты при p = 10.
Спаренные основания: 1-7 (71-65), 9-13 (22-26), 27-31 (39-43), 48-52 (59-63).
Псевдоузлы не учитываем.
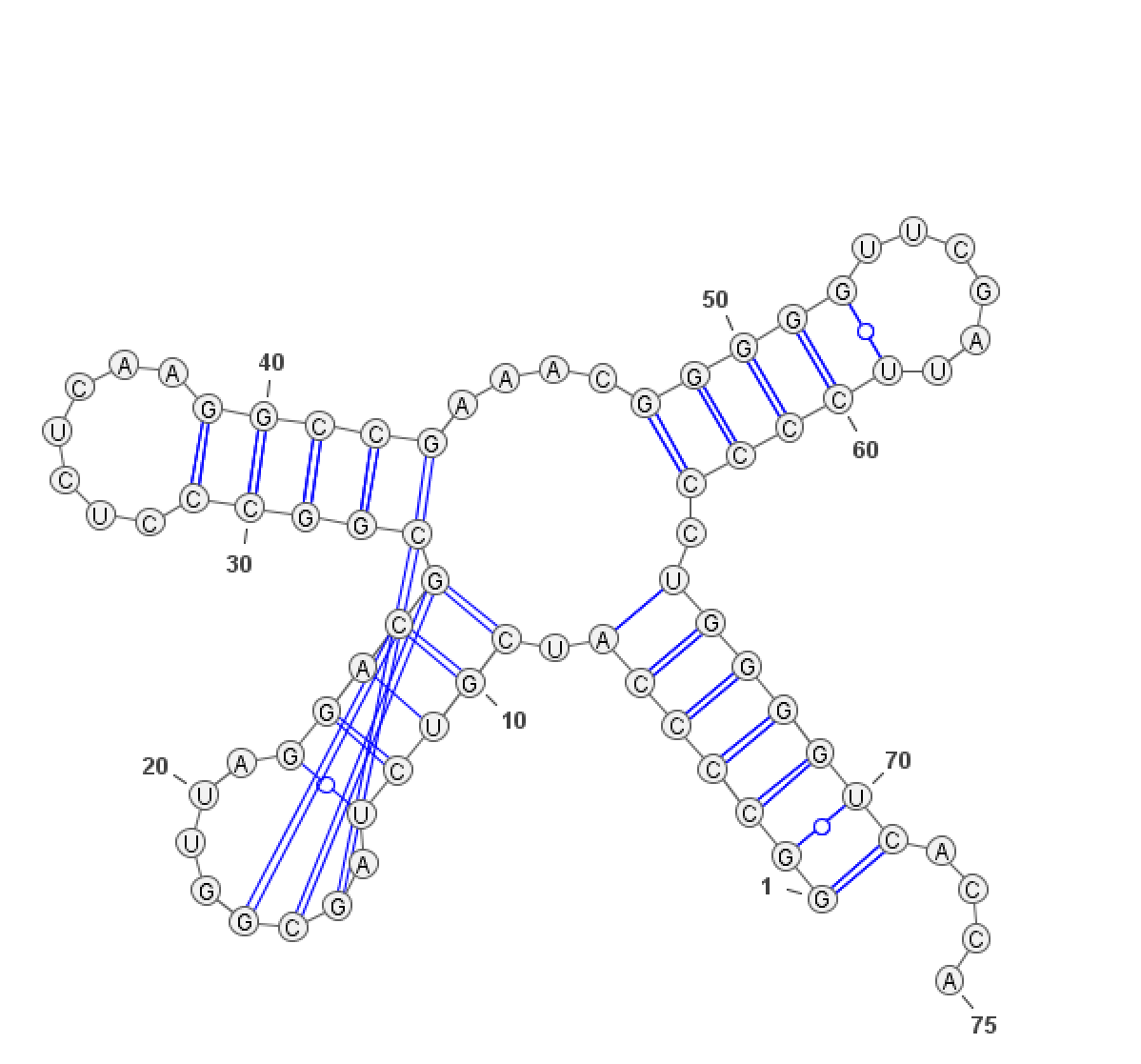
Таблица 2. Контакты разного типа в комплексе 1DDN.pdb
Участок структуры |
Позиции в структуре (по результатам find_pair) |
Результаты предсказания с помощью einverted |
Результаты предсказания по Zucker алгоритму |
Акцепторный стебель |
5'-501 - 507-3' |
предсказано 5 пар из 7 реальных |
предсказано 7 пар из 7 реальных |
D-стебель |
5'-510 - 512-3' |
предсказано 0 пар из 3 реальных |
предсказано 5 пар из 3 реальных |
T-стебель |
5'-549 - 553-3' |
предсказано 0 пар из 5 реальных |
предсказано 5 пар из 5 реальных |
Антикодоновый стебель |
5'-538 - 544-3' |
предсказано 0 пар из 7 реальных |
предсказано 5 пар из 7 реальных |
Общее число канонических пар нуклеотидов |
19 |
5 |
19 |