Комплексы ДНК-белок
Определение множеств в JMol
Зададим с помощью команды define следующие множества атомов в JMol:
- атомы кислорода 2'-дезоксирибозы (set1)
- атомы кислорода в остатке фосфорной кислоты (set2)
- атомы азота в азотистых основаниях (set3)
Теперь получим скрипт (файл по ссылке), вызов которого в JMol даст последовательное изображение всей структуры, только ДНК в проволочной модели, той же модели, но с выделенными шариками множеством атомов set1, затем set2 и set3.
Поиск ДНК-белковых контактов в структуре 1DDN
Будем считать полярными атомы кислорода и азота, а неполярными - атомы углерода, фосфора и серы. Назовем полярным контактом ситуацию, в которой расстояние между полярным атомом белка и полярным атомом ДНК меньше 3.5Å, неполярным контактом - пару неполярных атомов на расстоянии меньше 4.5Å. Средствами JMol найдём число различных контактов. Результаты приведены в таблице 1.
Таблица 1. Число контактов между ДНК и белком в 1DDN
| Контакты атомов белка с различными остатками | Полярные | Неполярные | Всего |
| 2'-дезоксирибозы | 13 | 47 | 60 |
| фосфорной кислоты | 45 | 129 | 174 |
| азотистых оснований со стороны большой бороздки | 9 | 21 | 30 |
| азотистых оснований со стороны малой бороздки | 0 | 3 | 3 |
Примечательно, что белок лежит только в больших бороздках ДНК (см. рис. 1). Это подтверждается таблицей 1. 3 атома, найденные как взаимодействующие с малой бороздкой, на самом деле лежат в большой, но достаточно глубоко в ней (на столько, что до атомов малой бороздки остаётся всего 4,5 Å). Почему с остатками фосфорной кислоты наблюдается много неполярных взаимодействий тоже понятно: это самая крайнаяя часть ДНК и расстояние берётся целых 4,5 Å (а не 3,5 как в случае с полярным взаимодействием).
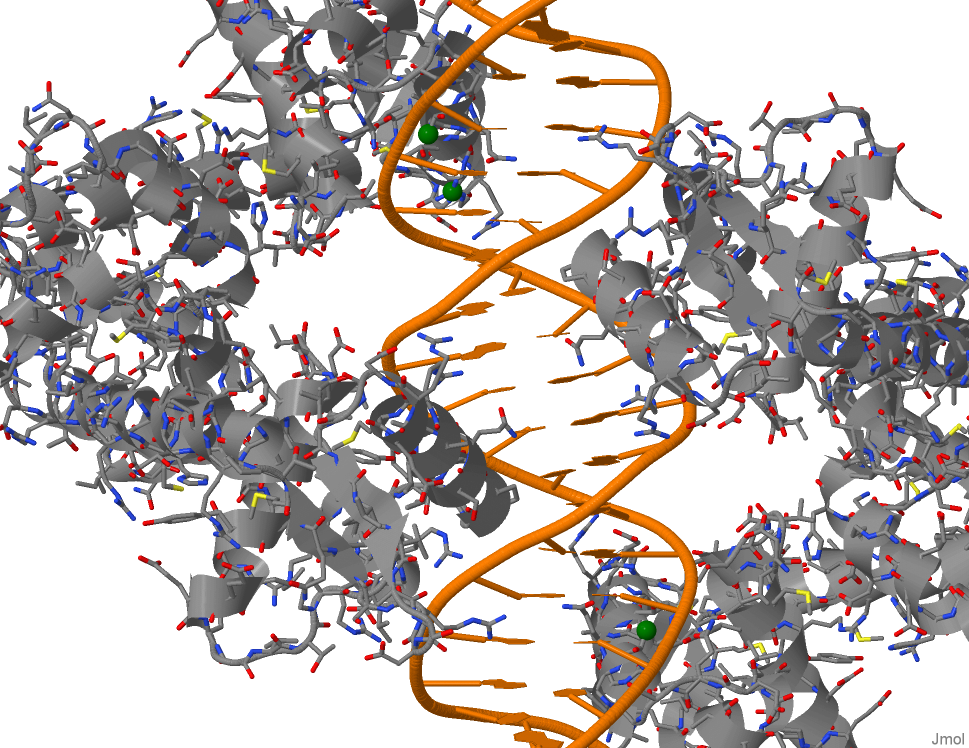
|
| Рис. 1 Структура 1DDN. Оранжевым цветом выделен ДНК-дуплекс, зелёными шариками - атомы, найденные взаимодействующими с малой бороздкой. |
Получение схемы ДНК-белковых взаимодействий
С помощью программы nuclplot, получим изобржение (рис.2), на котором отмечены ДНК-белковые взаимодействия.
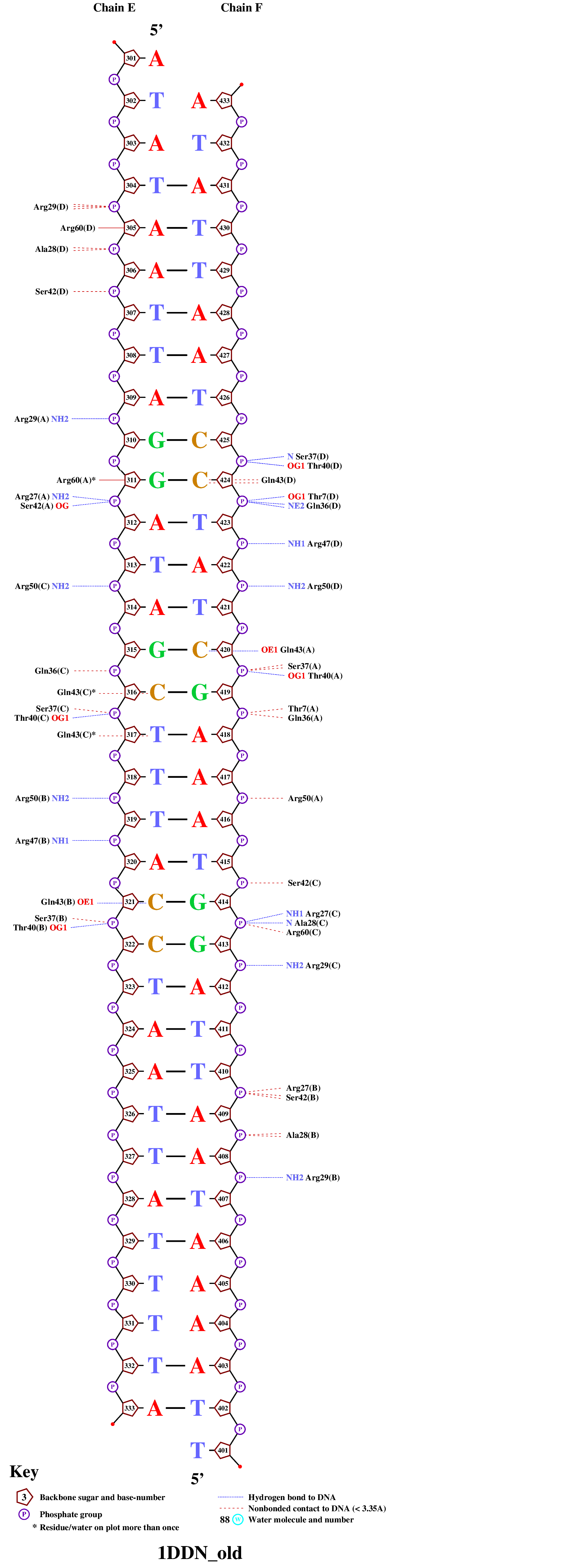
|
| Рис. 2 Изображение взаимодействий ДНК-белок |
Анализ результатов
Внимательно посмотрев на полученную схему, найдём аминокислотный остаток, образующий наибольшее число связей с ДНК. Это остаток глутамина-36 из цепи D, он образует две водородные связи с атомами фосфатной группы цитозина-424 (см. рис.3)
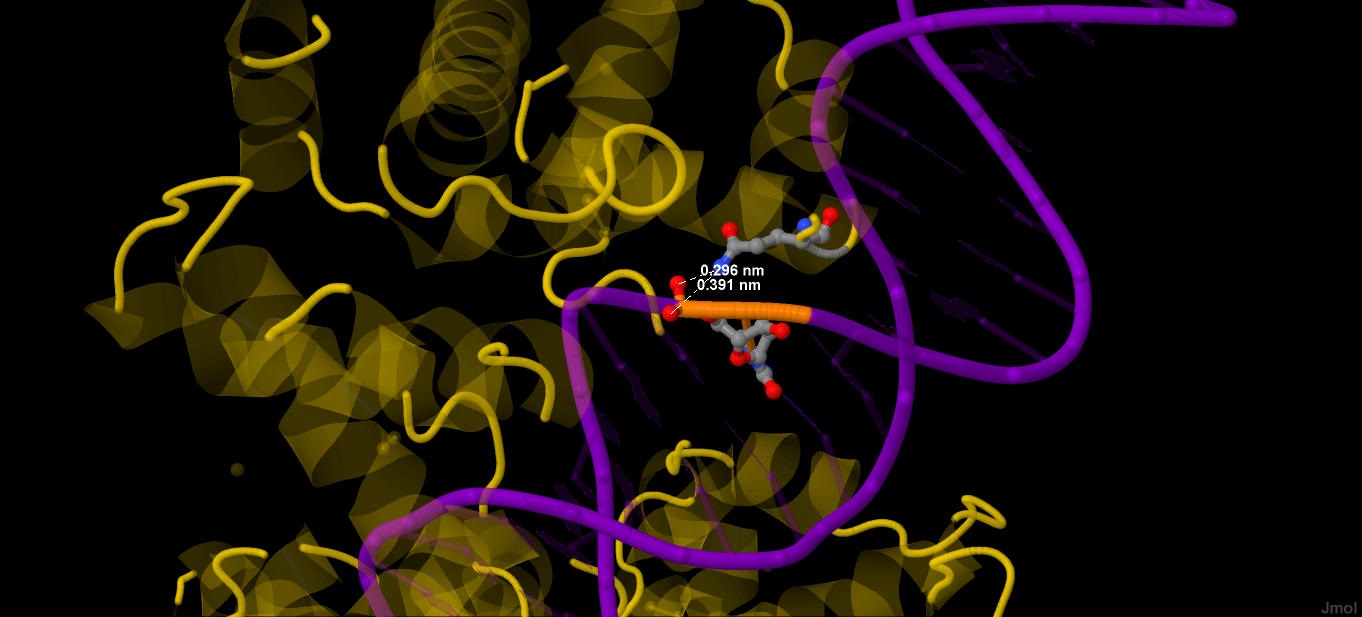
|
| Рис. 3 Взаимодействие атомов остатка глутамина-36 и фосфатной группы цитозина-424. Водородные связи показаны пунктиром, атомы окрашены в стандартные цвета. |
К наиболее важным аминокислотным остаткам для узнавания ДНК можно отнести тирозин-40 во всех четырёх цепях. Во-первых, он расположен в α-спирали, конформационно достаточно устойчивой части белка. Во-вторых, тирозин-40 из всех четырёх цепей образует водородные связи с ДНК, что говорит о консервативности этой связи. В-третьих, α-спирали, в которых расположены остатки тирозина-40 вдаются достаточно глубоко в большую бороздку. Такое специфическое расположение может играть роль в узнавании ДНК. Приведённые утверждения проиллюстрированы на рисунках 4 и 5.
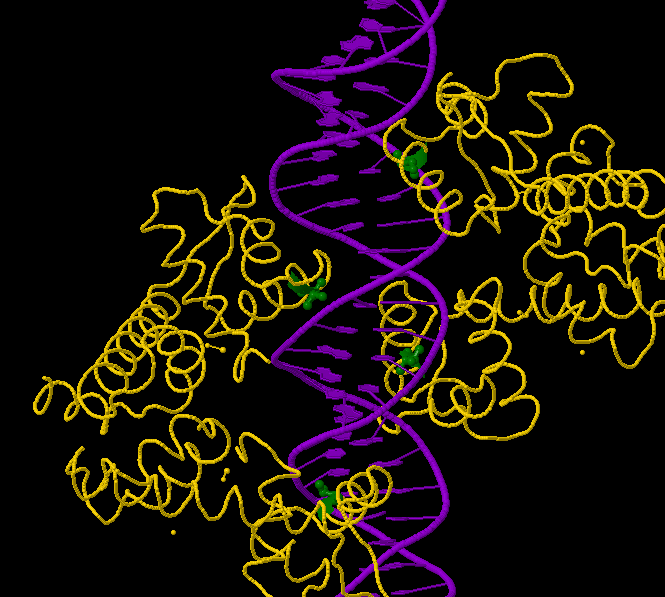
|
| Рис. 4 Расположение тирозинов-40 в комплексе ДНК-белок. ДНК выделена фиолетовым цветом, цепи белка - жёлтым, остатки тирозина-40, расположенные внутри больших бороздок - зелёным. |
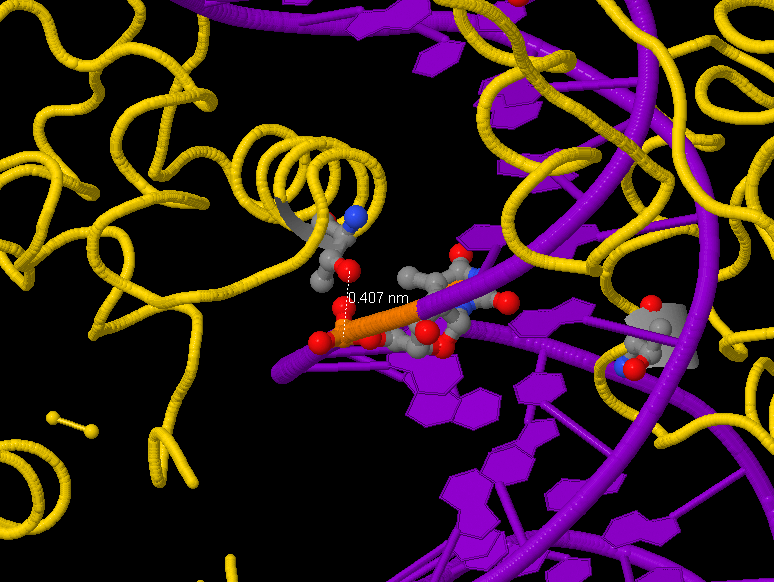
|
| Рис. 5 Взаимодействие атомов остатка глутамина-40 и фосфатной группы тимидила-317. Водородные связи показаны пунктиром, атомы окрашены в стандартные цвета. |
Предсказание вторичной структуры заданной тРНК
Подадим команде einverted на вход последовательность тРНК, с которой мы ранее имели дело. Используя параметры gap penalty: 4, minimum score treshold: 50, match score: 10, mismatch score: -4, на выходеполучаем следующее выравнивание:
1G59: Score 160: 23/28 ( 82%) matches, 15 gaps
1 g-g-ccccatcgtctagcgg-ttaggacgcggccc-tctcaag 39
| | ||||| | | || | | || | ||| | || |
73 cactggggt--c----ccccttagcttg-g--gggcaaag--c 42
Как видим, стебли тРНК не выделяются, просто первая половина последовательности выровнена со второй, что неверно. Однако акцепторный стебель всё же можно выделить.
Воспользуемся порталом mobyle@pasteur, чтобы получить предсказание инвертированных участков по алгоритму Зукера, сохраним лучшее предсказание (см. рис 6).
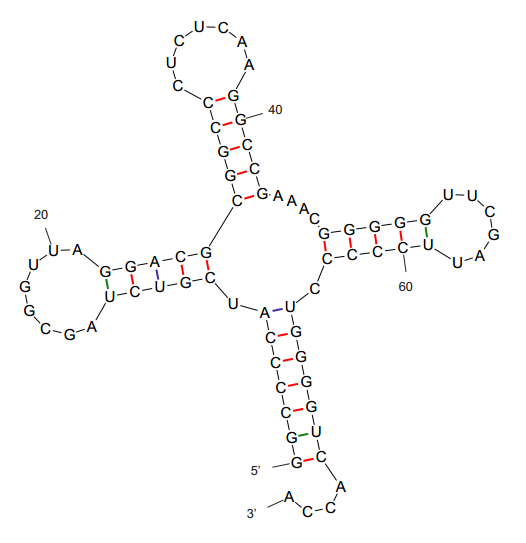
|
| Рис. 6 Вторичная структура тРНК из 1DDN, предсказанная по алгоритму Зукера. |
Сравнение полученных результатов приводится в таблице 2.
Таблица 2. Реальная и предсказанная вторичная структура тРНК из файла 1DDN.pdb
| Участок структуры | Позиции в структуре (по результатам find_pair) | Результаты предсказания с помощью einverted | Результаты предсказания по алгоритму Зукера |
| Акцепторный стебель | 5' 01-07 3' 3' 72-66 5' 7 пар | 6 пар из 7 | 7 пар из 7 |
| D-стебель | 5' 10-12 3' 3' 25-23 5' 3 пары | - | 3 пары из 3 + 2 лишних |
| T-стебель | 5' 26-32 3' 3' 44-38 5' 7 пар | - | 5 пар из 7 |
| Антикодоновый стебель | 5' 49-54 3' 3' 65-58 5' 7 пар | - | 5 пар из 7 |
| Общее число канонических пар нуклеотидов | 24 | 23 | 22 |