Реконструкция филогенетических деревьев
Филогения
С помощью сервиса taxonomy на сайте NCBI для бактерий, отобранных для выполнения предыдущего практикума, определим таксономическое положение. Полученные данные приведены в таблице 1.
Таблица 1. Таксономическое положение выбранных бактерий.
| Название вида | Мнемоника | Таксономическое положение |
| Bacillus anthracis | BACAN | Bacilli; Bacillales; Bacillaceae; Bacillus; Bacillus cereus group |
| Bacillus subtilis | BACSU | Bacilli; Bacillales; Bacillaceae; Bacillus |
| Clostridium tetani | CLOTE | Clostridia; Clostridiales; Clostridiaceae; Clostridium |
| Geobacillus kaustophilus | GEOKA | Bacilli; Bacillales; Bacillaceae; Geobacillus |
| Lactobacillus delbrueckii | LACDA | Bacilli; Lactobacillales; Lactobacillaceae; Lactobacillus |
| Lactococcus lactis | LACLM | Bacilli; Lactobacillales; Streptococcaceae; Lactococcus |
| Listeria monocytogenes | LISMO | Bacilli; Bacillales; Listeriaceae; Listeria |
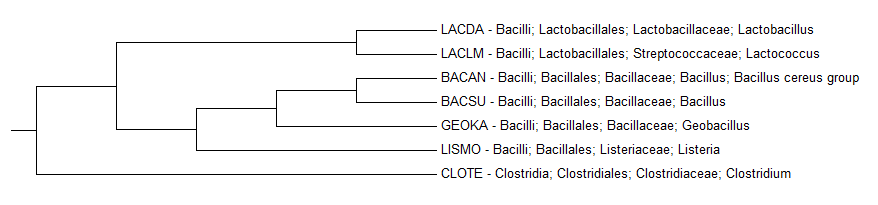
|
| Рис. 1 Филогенетическое дерево с указанием мнемоники и таксономии бактерий. |
Логично, что каждая ветвь на данном дереве, отделяет какой-нибудь таксон, например:
- Bacilli (класс) - {LACDA, LACLM, BACAN, BACSU, GEOKA, LISMO}
- Clostridia (класс) - {CLOTE}
- Lactobacillales (порядок) - {LACDA, LACLM}
- Bacillales (порядок) - {BACAN, BACSU, GEOKA, LISMO}
Более мелкие таксоны также отделены соответствующими им ветвями.
Создание и анализ выравнивания
Для реконструкции филогенетического дерева была выбрана одна иp предложенных функций белков - шаперонин (белок, имеющий внутреннюю полость и обеспечивающий правильно сворачивание других белков). Были получены последовательности шаперонинов для каждой из бактерий, и сделано их выравнивание с помощью программы muscle в JalView (см. рис. 2, jar-файл - здесь, fasta-файл - здесь). На полученном выравнивании можно определить диагностические позиции - те, по которым последовательность можно отнести к какому-либо таксону.
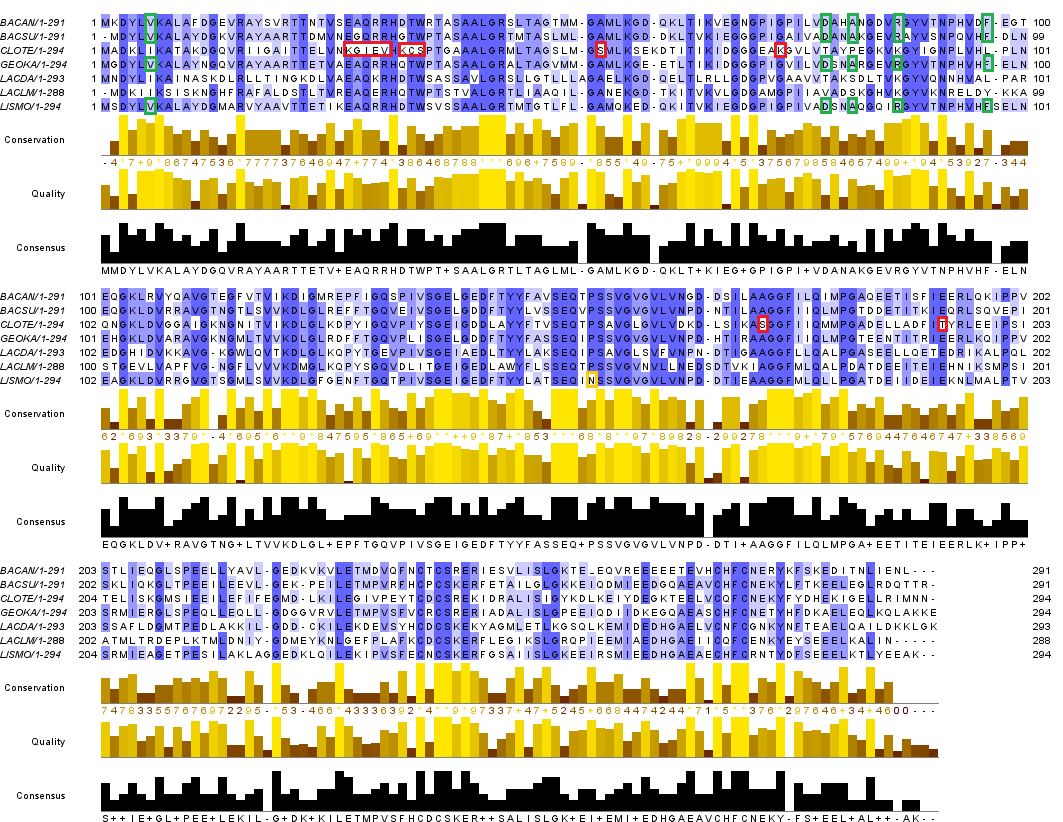
|
| Рис. 2 Выравнивание последовательностей шаперонинов из выбранных бактерий (окраска по проценту идентичности). Красными рамками выделены диагностические позиции для Clostridia, зелёными - для Bacillales, оранжевой - для Listeriaceae. |
Список диагностический позиций для нескольких таксонов:
- Clostridia - 28-32(KGIEV), 34-36(KCS), 54(S), 74(K), 174(S), 194(T);
- Bacillales - 6(V), 79(D), 82(A), 87(R), 97(F);
- Listeriaceae - 154(N).
Реконструкция филогенетического дерева
Алгоритмы Average distance и Neigbour Joining
По полученному выравниванию были построены филогенетические деревья всеми алгоритмами, имеющимися в JalView. Полученные деревья визуализированы с помощью программы Mega (см. таблицу 2).
Таблица 2. Филогенетические деревья, реконструированные по выравниванию различными алгоритмами.
| Метод | Average distance | Neigbour Joining |
| using % identity | 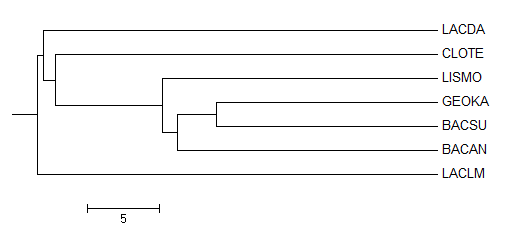
|
|
| using BLOSUM62 | 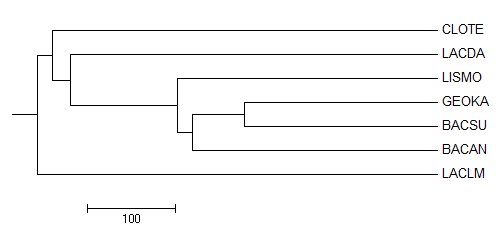
|
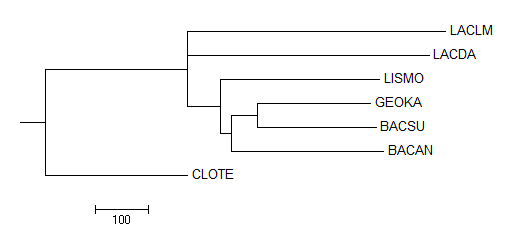
|
Ни одно из реконструированных деревьев не совпадает с праильным, в таблице 3 приведены данные о наличии различных ветвей в полученных деревьях.
Таблица 3. Ветви реконструированных деревьев.
| Ветвь | Правильное дерево | Neigbour Joining using BLOSUM62 | Average distance using BLOSUM62 | Neigbour Joining using % identity | Average distance using % identity | Maximum Parsimony |
| {BACAN, BACSU} vs {others} | + | - | - | - | - | - |
| {BACAN, BACSU, GEOKA} vs {others} | + | + | + | + | + | + |
| {BACAN, BACSU, GEOKA, LISMO} vs {others} | + | + | + | + | + | + |
| {LACDA, LACLM} vs {others} | + | - | + | - | - | - |
| {BACSU, GEOKA} vs {others} | - | + | + | + | + | - |
| {CLOTE, LACLM} vs {others} | - | + | - | - | - | - |
| {CLOTE, LACDA} vs {others} | - | - | - | - | - | + |
| {GEOKA, BACAN} vs {others} | - | - | - | - | - | + |
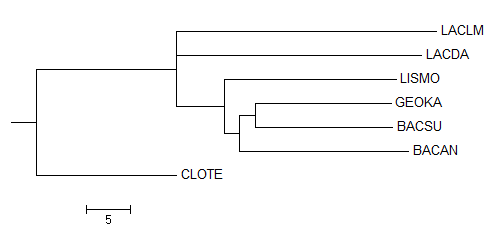
Алгоритм Maximum Parsimony
С помощью программы Mega по полученному ранее выравниванию было реконструировано дерево алгоритмом Maximum Parsimony и укоренено вручную (см. рис. 3). Данное дерево имеет две общих ветви с правильным (см. таблицу 3).
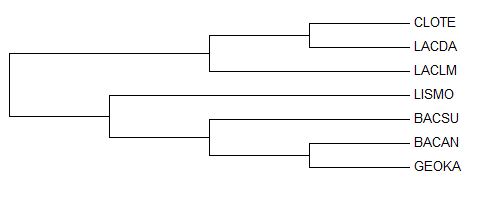
|
| Рис. 3. Дерево, реконструированное алгоритмом Maximum Parsimony. |
Несовпадение реконструированных деревьев с правильным вполне объяснимо. Реконструкция этих деревьев основывается на консервативности последовательностей всего одного белка. Такие деревья полезны для анализа эволюции одной последовательности. Правильное филогенетическое дерево составляется на основе выравниваний множества белков и РНК, понему можно судить об эволюции видов.