Для выполнения этого практикума мне было выдан файл с 6 хромосомой.
Сначала была произведена индексация референсной последовательности: hisat2-build chr6.fasta
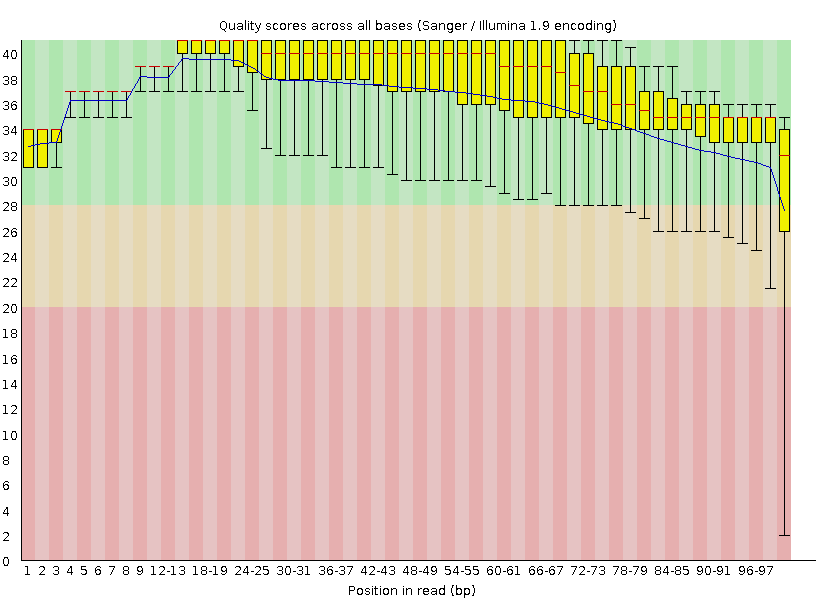
С помощью команды FastQC был проведен анализ качества одноконцевых чтений в формате fastq: fastqc chr6.fastq. Результат анализа можете
посмотреть здесь.
Изначально было 10289 ридов (длина 33-100).
Результаты анализа до использования Trimmomatic
Затем я сделала очистку чтений с помощью программы Trimmomatic. Командой
java -jar /nfs/srv/databases/ngs/suvorova/trimmomatic/trimmomatic-0.30.jar SE -phred33 chr6.fastq trim_chr6.fastq TRAILING:20 MINLEN:50
я отрезала с конца каждого чтения нуклеотиды с качеством ниже 20 и оставила только чтения длиной не меньше 50 нуклеотидов, а затем повторила
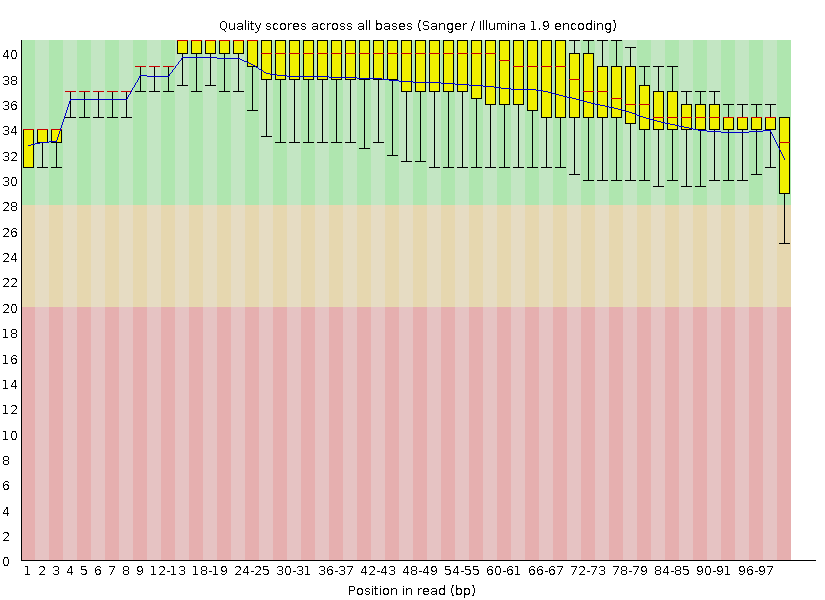
контроль качества. Оказалось, что чистка значительно улучшила качество последних позиций в риде.
Качество стало выше 25, но и до очистки качество было выше 30, за исключением последней позиции в риде.
Поэтому, хотя качество, конечно, и увеличилось, сложно говорить о том, насколько чистка была обоснованная. Результат здесь.
Осталось 10123 ридов, т.е. был удален 1,61% ридов.
Результаты анализа после использования Trimmomatic
Для картирования чтений было необходимо построить выравнивание прочтений и референса в формате .sam. Для этого
я использовала команду:
hisat2 --no-spliced-alignment --no-softclip -x /nfs/srv/databases/ngs/olga.shigal/ht2_chr6 -U /nfs/srv/databases/ngs/olga.shigal/trim_chr6.fastq -S chr6.sam,
Вывод команды:
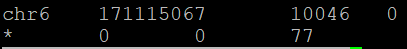
10123 reads; of these:
10123 (100.00%) were unpaired; of these:
77 (0.76%) aligned 0 times
10046 (99.24%) aligned exactly 1 time
0 (0.00%) aligned >1 times
99.24% overall alignment rate
Потом создала бинарный файл с расширением .bam: samtools view -b chr6.sam -o chr6.bam
Выравнивания с референсом я отсортировала по координате в референсе начала чтения (по умолчанию): samtools sort chr6.bam outsort
Проиндексировала отсортированный .bam файл командой: samtools index outsort.bam samtools idxstats outsort.bam > sumread.txt
Оказалось, что 10046 ридов откартировалось (99,2%), и 77 - не откартировалось, причем ни один рид не откартировался больше одного раза.
Таким образом, можно считать картирование качественным.
Результаты работы samtools idxstats
Поиск snp и инделей
Команда samtools mpileup -uf chr6.fasta -g -o polymor.bfc outsort.bam - создание файла с полиморфизмами.
Создание файла со списком отличий между референсом и чтениями bcftools call -cv polymor.bfc -o difer.vcf
Таблица 1. Описание полиморфизмов из файла .vcf
Полиморфизм
1
2
3
Координата
106961119
107016838
138192607
Тип полиморфизма
Замена
Вставка
Замена
Референс
T
CTTT
G
Чтение
C
CTTTTT
T
Глубина покрытия
6
36
29
Качество чтений
36.0297
217.468
28.0137
Среднее значение качества всех полиморфизмов получилось 117,6, медиана - 114. Для глубины покрытия среднее значение - 28, а медиана - 9, и
именно медиана лучше отображает реальную ситуацию, т.к. среднее значение смещено из-за немногих полиморфизмов глубина которых около 80 или даже больше.
Аннотация SNP
Перед началом работы с программой annovar, я конвертировала файл .vcf:convert2annovar.pl -format vcf4 difer.vcf -outfile difer.avinput
. Эта команда при выполнении сообщила следующее:"Всего 79 SNP, из них 50 транзиции и 29 трансверсии, и 6 инделей.
Далее производилась аннотация snp по разным базам.
Аннотация файла с snp по базе refgene:
annotate_variation.pl -out an_rg -build hg19 -dbtype refGene difer.avinput /nfs/srv/databases/annovar/humandb.old/
Из полученного файла an_rg.variant_function я узнала, что разделение на категории оказалось следующим: exonic (15 snp), intronic (64 snp),
UTR3 (5 snp). Гены, в которые попали SNP: AIM1, TNFAIP3, OPRM1. Из файла с экзонами узнаем о заменах:
line70 nonsynonymous SNV OPRM1:NM_001008505:exon4:c.A1206T:p.Q402H, chr6 154414446 154414446 A T hom 221.999 85
line71 synonymous SNV OPRM1:NM_001008505:exon4:c.A1323G:p.G441G, chr6 154414563 154414563 A G hom 221.999 84
line77 stopgain OPRM1:NM_001145286:exon4:c.C1231T:p.Q411X, chr6 154428666 154428666 C T het 225.009 26
Аннотация файла с snp по базе dbsnp:
annotate_variation.pl -filter -out ann_dbsnp -build hg19 -dbtype snp138 difer.avinput /nfs/srv/databases/annovar/humandb.old/
Из файла ann_dbsnp.hg19_snp138_dropped, в котором лежат snp c rs, узнаем, что таких 73 (из 79)
Аннотация файла с snp по базе 1000 genomes:
annotate_variation.pl -filter -out ann_1000g -build hg19 -dbtype 1000g2014oct_all difer.avinput /nfs/srv/databases/annovar/humandb.old/
Из этой аннотации мы можем узнать частоту встречаемости каждого полиморфизма. Всего было аннотировано 73 полиморфизма. Средняя частота каждого snp составляет 45% (медиана 38,6%), причем самый редкий snp 0,659%,
а самый частый 98,78%
Аннотация файла с snp по базе Gwas (region-based annotation):
annotate_variation.pl -regionanno -out ann_gwas -build hg19 -dbtype gwasCatalog difer.avinput /nfs/srv/databases/annovar/humandb.old/
В Gwas есть информация по 4 snp:
gwasCatalog Name=Stroke chr6 106987370 106987370 A C hom 221.999 88
gwasCatalog Name=Systemic lupus erythematosus chr6 138195723 138195723 C G het 65.0073 9
gwasCatalog Name=Rheumatoid arthritis,Systemic lupus erythematosus chr6 138196066 138196066 T G het 225.009 46
gwasCatalog Name=Coronary heart disease chr6 154414563 154414563 A G hom 221.999 84
Аннотация файла с snp по базе Clinvar (filter-based annotation):
annotate_variation.pl -filter -out an_clinvar -build hg19 -dbtype clinvar_20150629 difer.avinput /nfs/srv/databases/annovar/humandb.old/
Аннотация дала информацию по одном snp:
clinvar_20150629 CLINSIG=untested; CLNDBN=not_specified; CLNREVSTAT=no_assertion_provided; CLNACC=RCV000122149.1; CLNDSDB=MedGen; CLNDSDBID=CN169374 chr6 138196066 138196066 T G het 225.009 46
Клиническая аннотация
Посмотрим на содержимое файла, полученного после аннотации по Gwas:
Stroke (Инсульт) chr6 106987370 106987370 A C hom 221.999
Systemic lupus erythematosus(Системная красная волчанка) chr6 138195723 138195723 C G het 65.0073
Rheumatoid arthritis (Ревматоидный артрит),Systemic lupus erythematosus chr6 138196066 138196066 T G het 225.009
Coronary heart disease (Ишемическая болезнь сердца) chr6 154414563 154414563 A G hom 221.999
Вывод
Возможно, аннотация дала недостаточную информацию о всех полиморфизмах пациента, но позволила узнать о связи 4 snp с заболеваниями.