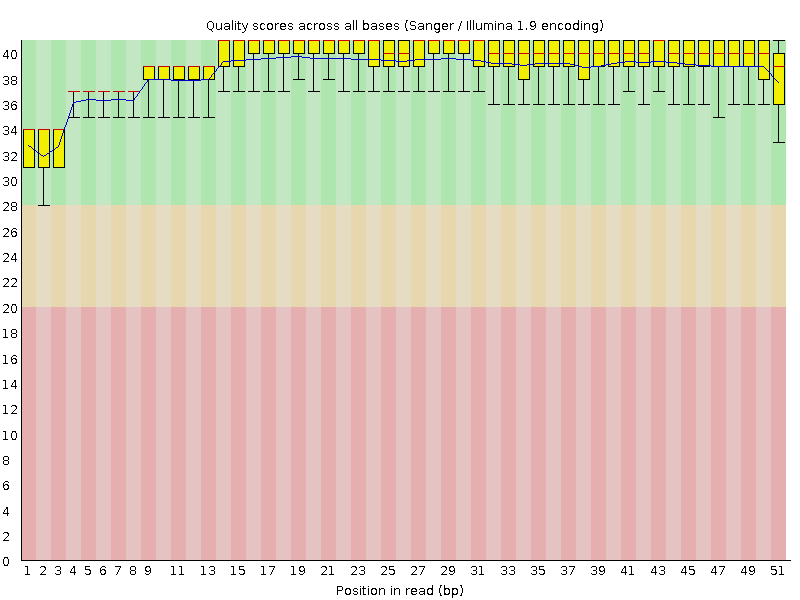
Результаты анализа до использования Trimmomatic
| Команда | Что делает |
| fastqc chr6.1.fastq | Контроль качества чтений в формате .fastq. Результат |
| java -jar /nfs/srv/databases/ngs/suvorova/trimmomatic/trimmomatic-0.30.jar SE -phred33 chr6.1.fastq trim_chr6.1.fastq TRAILING:20 MINLEN:50 TrimmomaticSE: Started with arguments: -phred33 chr6.1.fastq trim_chr6.1.fastq TRAILING:20 MINLEN:50 | Очистка чтений с помощью Trimmomatic. Отрезает с конца каждого чтения нуклеотиды с качеством ниже 20, оставляет только чтения длиной не меньше 50 нуклеотидов. |
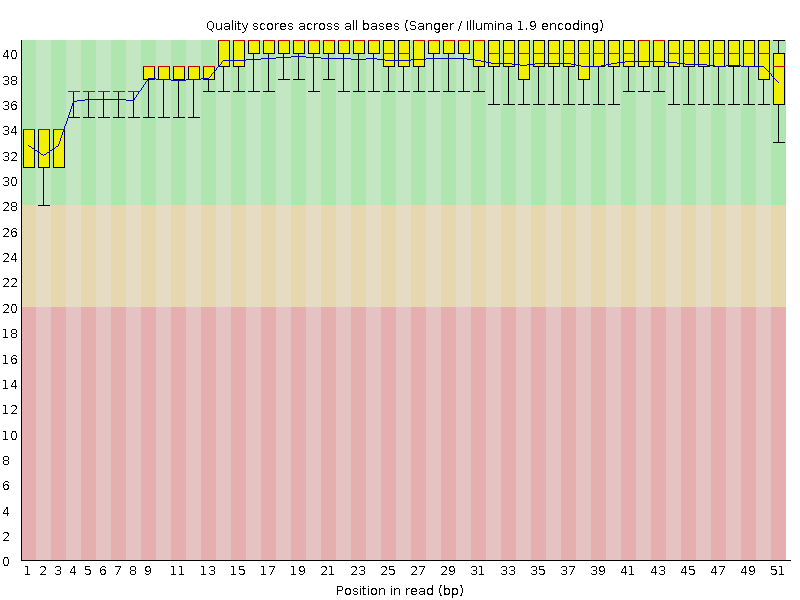
| fastqc trim_chr6.1.fastq | Анализ качества после чистки. Результат |
| hisat2 --no-softclip -x /nfs/srv/databases/ngs/olga.shigal/ht2_chr6 -U /nfs/srv/databases/ngs/olga.shigal/pr12/chr6.1.fastq -S chr6.sam | Выравнивание прочтений и референса в формате .sam. Из команды прошлого прака был убран параметр --no-spliced-alignment, из-за того, что теперь мы работаем с РНК-транскриптомами, из которых вырезаны интроны. |
| samtools view -b chr6.sam -o chr6.bam | Перевод выравнивания чтений в бинарный файл |
| samtools sort chr6.bam outsort | Сортировка выравнивания с референсом по координате в референсе начала чтения (по умолчанию) |
| htseq-count -f bam -s no -i gene_id -m union outsort.bam /nfs/srv/databases/ngs/Human/rnaseq_reads/gencode.v19.chr_patch_hapl_scaff.annotation.gtf > htseqc_results | Подсчет ридов для каждого из генов. -f: формат входного файла(bam), -s: цепь выравнивания чтений (no = неопределенность направления), -i: feature ID (по-умолчанию нужный) |
| grep -wv 0 htseqc_results > htseq.txt | Отделяем строчки с генами, у которых были риды. |
54019 reads; of these:
54019 (100.00%) were unpaired; of these:
9239 (17.10%) aligned 0 times
44780 (82.90%) aligned exactly 1 time
0 (0.00%) aligned >1 times
82.90% overall alignment rateЗначит, всего на геном было откартировано 44780 чтений.
ENSG00000156508.13 44605
__no_feature 175
__not_aligned 9239Оказалось, что неоткартированных ридов было 9239(согласуется с выводом hisat2), для 175 ридов не были определены границы генов, и 44605 ридов легли на ген ENSG00000156508.13. Данный ген кодирует эукариотический фактор элонгации трансляции 1 субъединицу α-1 (eEF1A1).