Практикум 11. Ресеквенирование. Поиск полиморфизмов у человека.
В данном практикуме были найдены и описаны полиморфизмы в экзоме 15 хромосомы человека.
Использованные команды для вызова программ
Описание использованных команд приведено в таблице 1.
Команда |
Описание |
hisat2-build chr15.fasta chr15 |
Индексирует последовательность ДНК |
fastqc chr15.fastq |
Оценивает качество прочтений (вывод: html-файл) |
java -jar /nfs/srv/databases/ngs/suvorova/trimmomatic/trimmomatic-0.30.jar SE -phred33 chr15.fastq chr15_trimmed.fastq TRAILING:20 MINLEN:50 |
Программа Trimmomatic. Очищает чтения по заданным параметрам |
hisat2 -x chr15 -U chr15_trimmed.fastq --no-spliced-alignment --no-softclip -S map.sam |
Строит выравнивание прочтений и референса в sam-формате |
samtools view map.sam -b -o map.bam |
Переводит sam-файл в бинарный формат |
samtools sort map.bam map_sorted |
Сортирует прочтения по координате начала чтения в референсе |
samtools index map_sorted.bam |
Индексирование бинарного файла |
samtools flagstat map_sorted.bam |
Показывает статистику откартированных чтений |
samtools mpileup -uf chr15.fasta -o pol.bcf map_sorted.bam |
Создание файла с полиморфизмами в бинарном формате |
bcftools call -cv pol.bcf -o pol.vcf |
Создание vcf-файла со списком отличий между референсом и прочтениями |
vcftools --vcf pol.vcf --remove-indels --recode --out snp |
Удаляет индели из прочтений |
convert2annovar.pl -format vcf4 snp.recode.vcf > snp.avinput |
Перевод vcf-файла в annovar-формат (.avinput) |
annotate_variation.pl -out snp_annotated -build hg19 -dbtype refGene snp.avinput /nfs/srv/databases/annovar/humandb/ |
Аннотирует полученные SNP по базе данных RefGene |
annotate_variation.pl -filter -out snp_annotated -build hg19 -dbtype snp138 snp.avinput /nfs/srv/databases/annovar/humandb.old/ |
Аннотирует полученные SNP по базе данных dbsnp |
annotate_variation.pl -filter -out snp_annotated -build hg19 -dbtype 1000g2014oct_all snp.avinput /nfs/srv/databases/annovar/humandb.old/ |
Аннотирует полученные SNP по базе данных 1000 genomes |
annotate_variation.pl -regionanno -out snp_annotated -build hg19 -dbtype gwasCatalog snp.avinput /nfs/srv/databases/annovar/humandb.old/ |
Аннотирует полученные SNP по базе данных Gwas |
annotate_variation.pl -filter -out snp_annotated -build hg19 -dbtype clinvar_20150629 snp.avinput /nfs/srv/databases/annovar/humandb.old/ |
Аннотирует полученные SNP по базе данных Clinvar |
Получение чтений экзома
Файлы:
chr15.fastq - исходные чтения
chr15_fastqc.html - html-отчёт качества исходных чтений
chr15_trimmed.fastq - триммированные чтения
chr15_trimmed_fastqc.html - html-отчёт качества триммированных чтений
В исходном fastq-файле находилось 5068 чтений. После триммирования осталось 4946 чтений. Сравнение качества чтений приведено на рисунке 1. Также на рисунке 2 приведены распределения качества прочтений до и после триммирования.
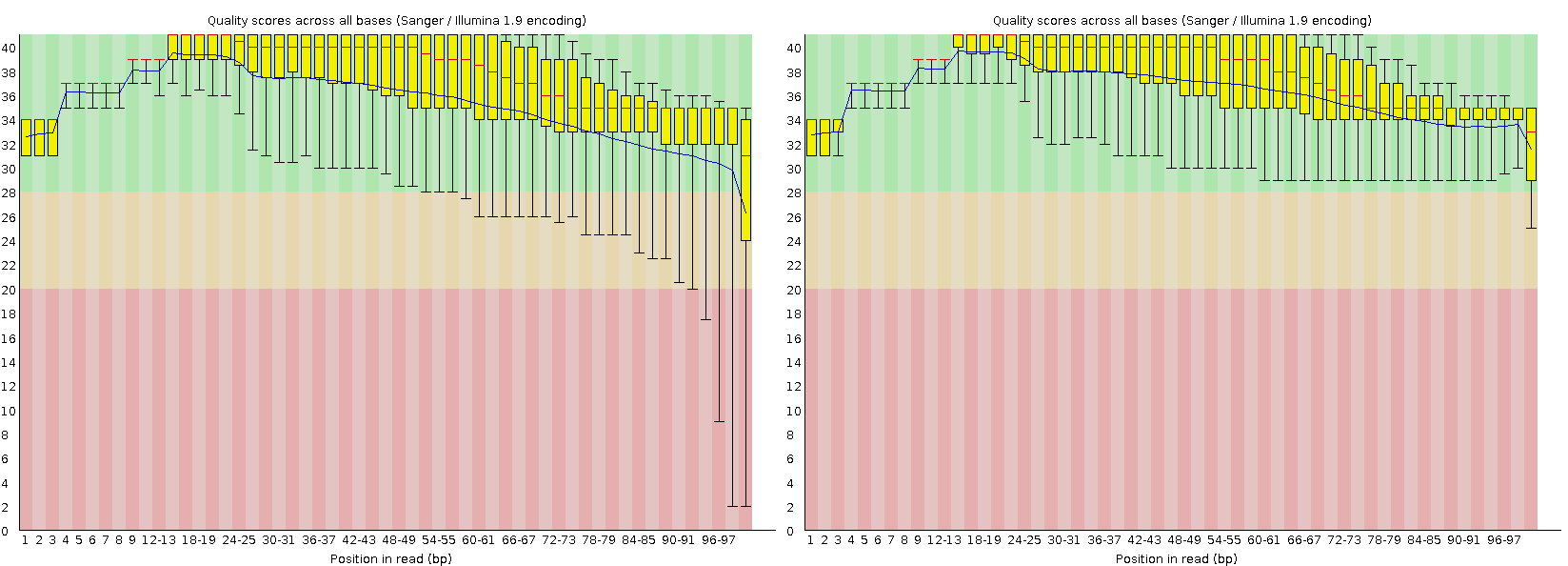
Рисунок 1. Сравнения качества прочтений до (слева) и после (справа) триммирования. Качество было улучшено за счёт удаления последовательностей длиной менее 50 п.н., а также последовательностей качеством менее 20.
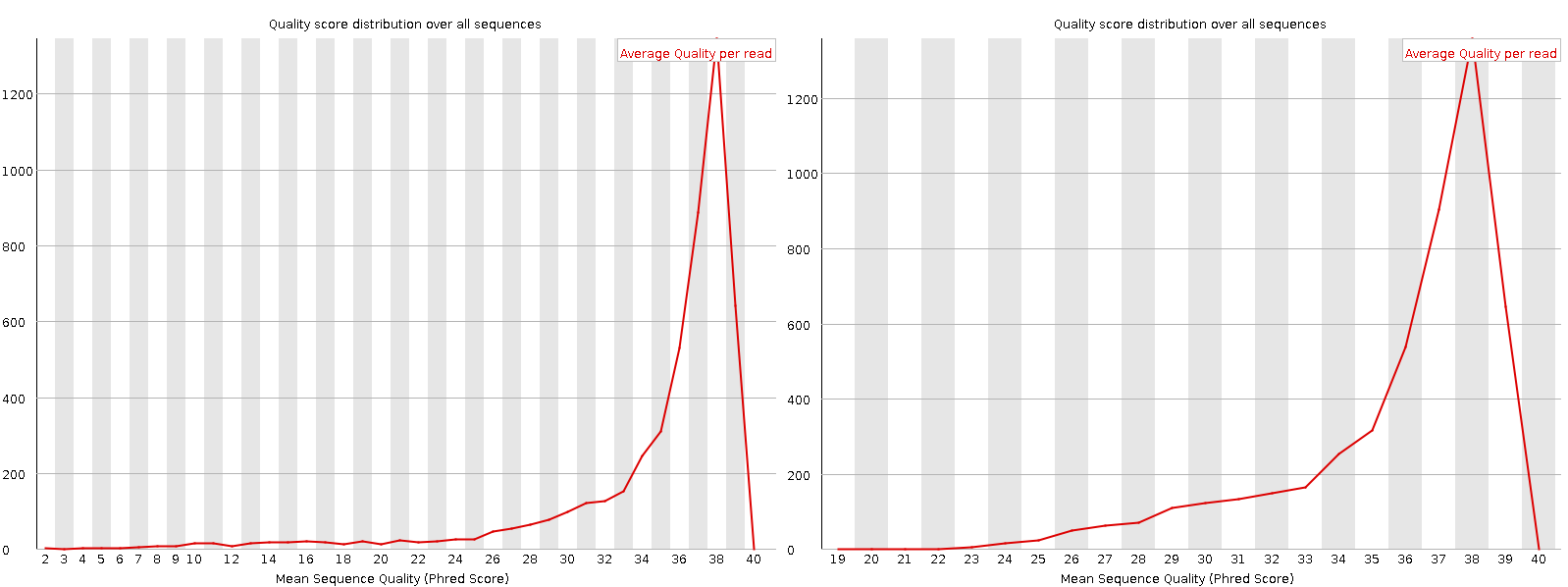
Рисунок 2. График распределения качества до (слева) и после (справа) триммирования прочтений. В ходе триммирования были удалены чтения длиной меньше 20, что подтверждается этим графиком.
В итоге триммированием было удалено 2.43% чтений от их исходного числа. При этом среднее качество прочтений слабо изменилось (рисунок 2), однако триммирование можно считать оправданным, так как оно показало явное улучшение качества прочтений.
Картирование триммированных чтений экзома на геном
Для начала была проиндексирована референсная последовательность (15 хромосома человека). Затем было проведено картирование чтений на референсную последовательность программой HISAT2. Вывод stdout:
4946 reads; of these:
4946 (100.00%) were unpaired; of these:
11 (0.22%) aligned 0 times
4935 (99.78%) aligned exactly 1 time
0 (0.00%) aligned >1 times
99.78% overall alignment rate
Не были выровнены 11 чтений из 4946, что составляет 0.22%. Остальные чтения выровнены ровно 1 раз. В итоге картировано 99.78% чтений, что позволяет говорить о хорошем картировании чтений на геном.
Поиск полиморфизмов в выравнивании чтения и генома
Файлы:
map.sam - выравнивание прочтений и референса
pol.vcf - отличия между чтениями и референсом
Был проиндексирован предварительно переведенный в бинарный формат и отсортированный файл с выравниванием чтений на геном. Затем был создан bcf-файл, преобразованый после в vcf-файл со списком отличий между чтениями и референсом. Всего получено 89 полиморфизмов: 87 SNP и 2 инделя. Данные о распределении качества и глубины покрытия полиморфизмов приведены на рисунке 3 (получено с помощью Excel). Описание трёх выбранных полиморфизмов приведено в таблице 2.
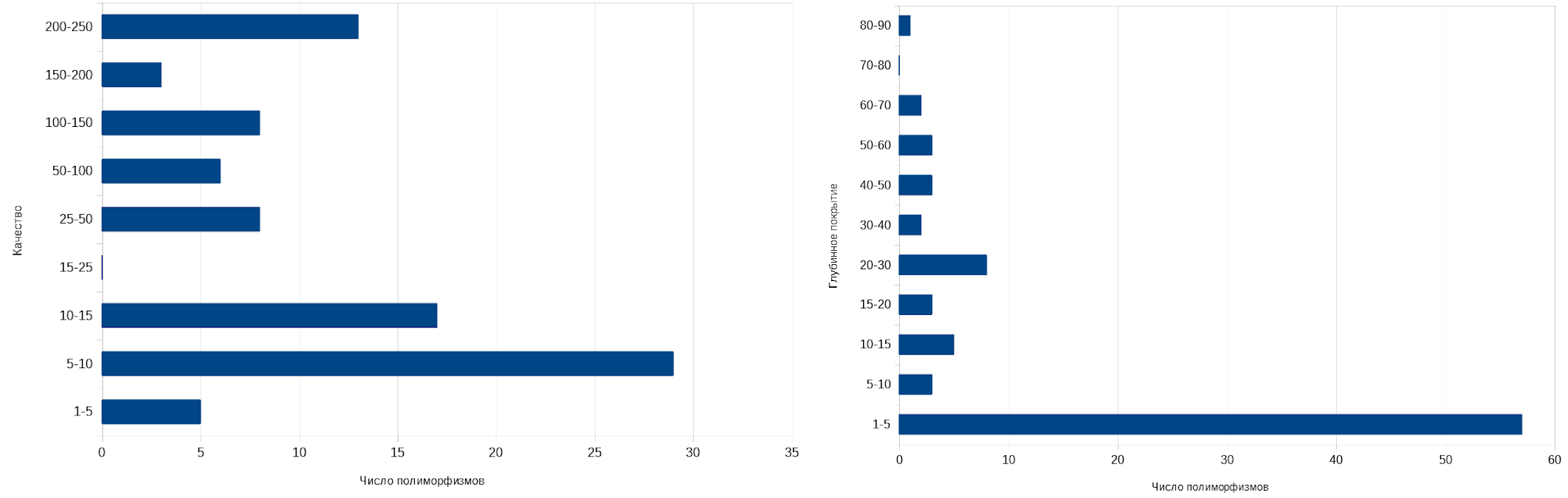
Рисунок 3. Распределение качества и глубинного покрытия у найденных полиморфизмов.
Преобладают полиморфизмы с низким качеством и небольшим глубинным покрытием. Тем не менее покрытие и качество можно считать удовлетворительными.
Координата |
Тип |
Референс |
Чтение |
Глубина покрытия |
Качество чтения |
58706050 |
Замена |
C |
A |
1 |
10.4247 |
58840482 |
Замена |
T |
C |
89 |
225.009 |
58853212 |
Делеция |
GACACAC |
GACAC |
- |
217.468 |
На рисунке 4 графически отображена информация об описанных полиморфизмах.
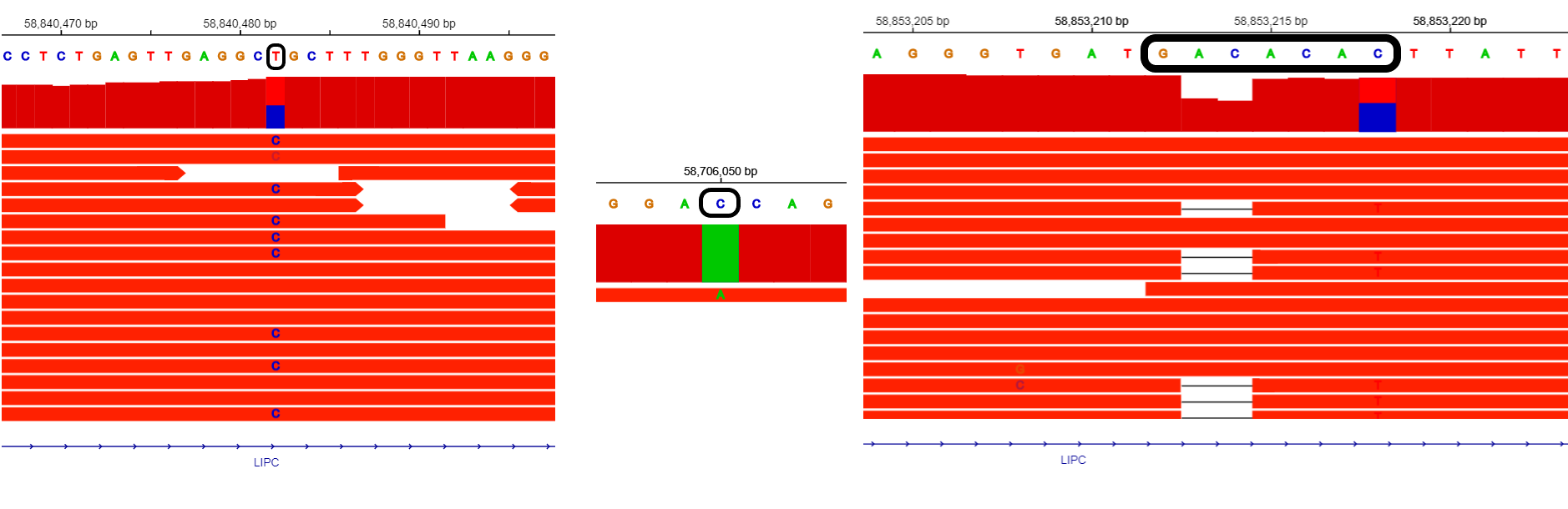
Рисунок 4. Изображение полиморфизмов из таблицы 2, полученное программой IGV. Для замены T-C более половины ридов не попали на изображение.
Аннотация SNP по базам данных RefGene, dbSNP, 1000 genomes, GWAS, Clinvar
Для аннотирования из vcf-файла были удалены индели, затем файл был переведён в формат annovar (snp.avinput). Описанными выше командами были получены аннотации SNP по 5 базам данных.
Файлы с полученными аннотациями:
RefGene: snp_annotated.variant_function ||
snp_annotated.exonic_variant_function
dbSNP: snp_annotated.hg19_snp138_dropped ||
snp_annotated.hg19_snp138_filtered
1000 genomes: snp_annotated.hg19_ALL.sites.2014_10_dropped ||
snp_annotated.hg19_ALL.sites.2014_10_filtered
GWAS: snp_annotated.hg19_gwasCatalog
Clinvar: snp_annotated.hg19_clinvar_20150629_filtered || пустой _dropped-файл
RefGene делит 87 SNP на 6 групп: intronic (60), ncRNA_intronic (10), exonic (12), intergenic (1), upstream (3), UTR3 (1). Гены с SNP: AQP9, LIPC, HMG20A, ACAN. Все нуклеотидные (с.) и аминокислотные (p.) замены перечислены в файле exonic_variant_function. Читаемый вариант:
synonymous LIPC:NM_000236:exon2:c.C264T:p.H88H, 58830707 58830707 C T het 225.009 . synonymous LIPC:NM_000236:exon4:c.G465T:p.V155V, 58834741 58834741 G T het 201.009 . synonymous LIPC:NM_000236:exon5:c.A591G:p.G197G, 58837957 58837957 A G hom 221.999 . nonsynonymous LIPC:NM_000236:exon5:c.A644G:p.N215S, 58838010 58838010 A G hom 221.999 . nonsynonymous LIPC:NM_000236:exon7:c.C1068A:p.F356L, 58853079 58853079 C A het 225.009 . synonymous LIPC:NM_000236:exon7:c.A1098G:p.T366T, 58853109 58853109 A G het 225.009 . nonsynonymous ACAN:NM_001135:exon3:c.C306A:p.D102E, 89382129 89382129 C A het 95.0077 . synonymous ACAN:NM_001135:exon7:c.C1221T:p.V407V, 89388905 89388905 C T het 184.009 . synonymous ACAN:NM_001135:exon9:c.C1623A:p.P541P, 89391160 89391160 C A hom 221.999 . synonymous ACAN:NM_001135:exon10:c.T1809C:p.A603A, 89392745 89392745 T C hom 221.999 . synonymous ACAN:NM_001135:exon10:c.C1968T:p.Y656Y, 89392904 89392904 C T het 129.008 . nonsynonymous ACAN:NM_001135:exon10:c.G2003A:p.R668Q, 89392939 89392939 G A het 129.008 .
Например, в первой строке несмотря на замену цитозина на тимин аминокислота осталась та же - гистидин.
По данным аннотации dbSNP имеется 75 SNP с rs.
По данным аннотации 1000 genomes о частоте SNP среднее частоты - 0,492, медиана - 0,478 (подсчитано при помощи Excel).
GWAS предоставляет клиническую аннотацию. Выяснилось, что 5 полиморфизмов связаны с ростом, холестерином высокой плотности, биохимией крови и диабетом 2 типа.
Отсутствуют результаты по Clinvar.