Для выполнения задания была взята итоговая последовательность, полученная в результате выполнения практикума 6. Был запущен алгоритм blastn (Somewhat similar sequences) по базе данных Nucleotide collection (nr/nt)
с параметрами по умолчанию.
Лучшие результаты blastn
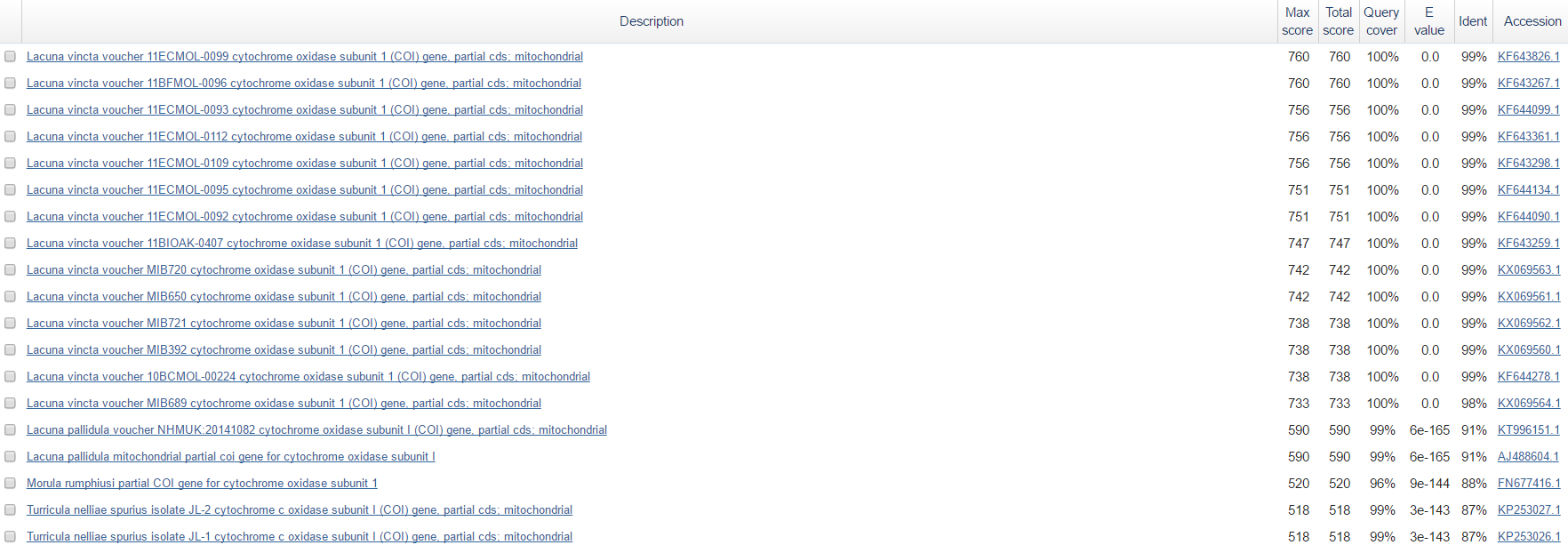 Отсеквенированный участок является частью митохондриального гена COI, в котором закодирован белок 1-ой субъединицы цитохром с-оксидазы. Цитохром с-оксидаза
Отсеквенированный участок является частью митохондриального гена COI, в котором закодирован белок 1-ой субъединицы цитохром с-оксидазы. Цитохром с-оксидаза
|
Цитохром с-оксидаза (цитохромоксидаза) — терминальная оксидаза аэробной дыхательной цепи переноса электронов, которая катализирует
перенос электронов с цитохрома с на кислород с образованием воды. Цитохромоксидаза присутствует во внутренней мембране митохондрий
всех эукариот, где её принято называть комплекс IV, а также в клеточной мембране многих аэробных бактерий.
Комплекс IV последовательно окисляет четыре молекулы цитохрома с и, принимая четыре электрона, восстанавливает O2 до H2O.
При восстановлении O2 четыре H+ захватываются из митохондриального матрикса для образования двух молекул H2O, а ещё четыре H+
активно перекачиваются через мембрану. Таким образом, цитохромоксидаза вносит свой вклад в создание протонного градиента для
синтеза АТФ и является частью пути окислительного фосфорилирования. Кроме того, этот мультибелковый комплекс играет ключевую
роль в регуляции активности всей дыхательной цепи и производстве энергии эукариотической клеткой.
Комплекс IV из митохондрий млекопитающих и птиц состоит из 13 белковых субъединиц, три из которых обладают каталитической
активностью, связывают кофакторы и кодируются генами митохондрий. Остальные десять субъединиц закодированы в ДНК ядра.
Три большие субъединицы комплекса (I—III), гомологичные бактериальным, несут на себе все необходимые кофакторы и
осуществляют основные реакции катализа, связанные, в том числе, и с переносом протонов.[1]
Для определения таксономии прочтенной последовательности было построено выравнивание первых 6-ти находок blastn (seqdump_all.txt).
Все 6 отсеквенированных участков гена, которые были найдены программой, принадлежат представителям вида Lacuna vincta-
брюхоногому моллюску, обитающему преимущественно на водорослях в литоральной и сублиторальной зонах Атлантики, Тихого Океана
и Северной Европы.[2] |
 |
Видно, что все последовательности практически идентичны и имеют несколько замен в пределах вида. Концевые участки последней
последовательности обрезаны, возможно, из-за нечитаемости хроматограммы, полученной при секвенировании этого участка гена.
Первые 14 находок blastn принадлежат Lacuna vincta, следующие 2 относятся уже к другому виду этого же рода: Lacuna pallidula.

Рис.1 Выравнивание, раскрашенное по схеме Nucleotide с добавлением LOGO.
Ссылка на JalView project
Таксономическое положение организма: Eukaryota; Opisthokonta; Metazoa; Eumetazoa; Bilateria; Protostomia; Lophotrochozoa; Mollusca; Gastropoda;
Caenogastropoda; Hypsogastropoda; Littorinimorpha; Littorinoidea; Lacunidae; Lacuna; Lacuna vincta.
Сравнение списков находок разных алгоритмов BLAST
*Поиск проводился против базы данных Reference genomic sequences из-за реорганизации сайта и невозможности ограничить таксономически поиск по базе Nucleotide collection.
Для выполнения задания необходимо было сравнить списки находок по нуклеотидной последовательности тремя алгоритмами BLAST: blastn, megablast и discontiguous megablast.
Чтобы результат сравнения был показателен, необходимо было таксономически ограничить область поиска. Поиски при использовании megablast по роду(Lacuna), семейству(Lacunidae), надсемейству(Littorinoidea),
подотряду(Littorinimorpha) и классу(Gastropoda) давали слишком мало находок (max по классу 8), а при использовании алгоритмов blastn и discontiguous megablast количество находок превышало 100. Для оптимизации
количества находок в среднем поиск в итоге был ограничен таксоном Hypsogastropoda-промежуточным звеном между классом и надотрядом(предположительно, отрядом, хотя в таксономии NCBI не обозначено название этого
таксона). В этом случае количество находок, найденных с помощью megablast, равно 8, с помощью blastn и discontiguous megablast равно 43.
| Параметры запуска BLAST | ||
|---|---|---|
| | Max Target Sequences | Expect Threshold |
Word Size | Max matches | Match/Mismatch Scores | Gap Costs |
| megablast | 250 | 10 | 28 |
0 | 1, -2 | Linear |
| discontiguous megablast | 250 | 10 |
11 | 0 | 2, -3 | Existence:5, Extention: 2 |
| blastn | 250 | 10 | 11 |
0 | 2, -3 | Existence:5, Extention: 2 |
Результаты выдачи megablast
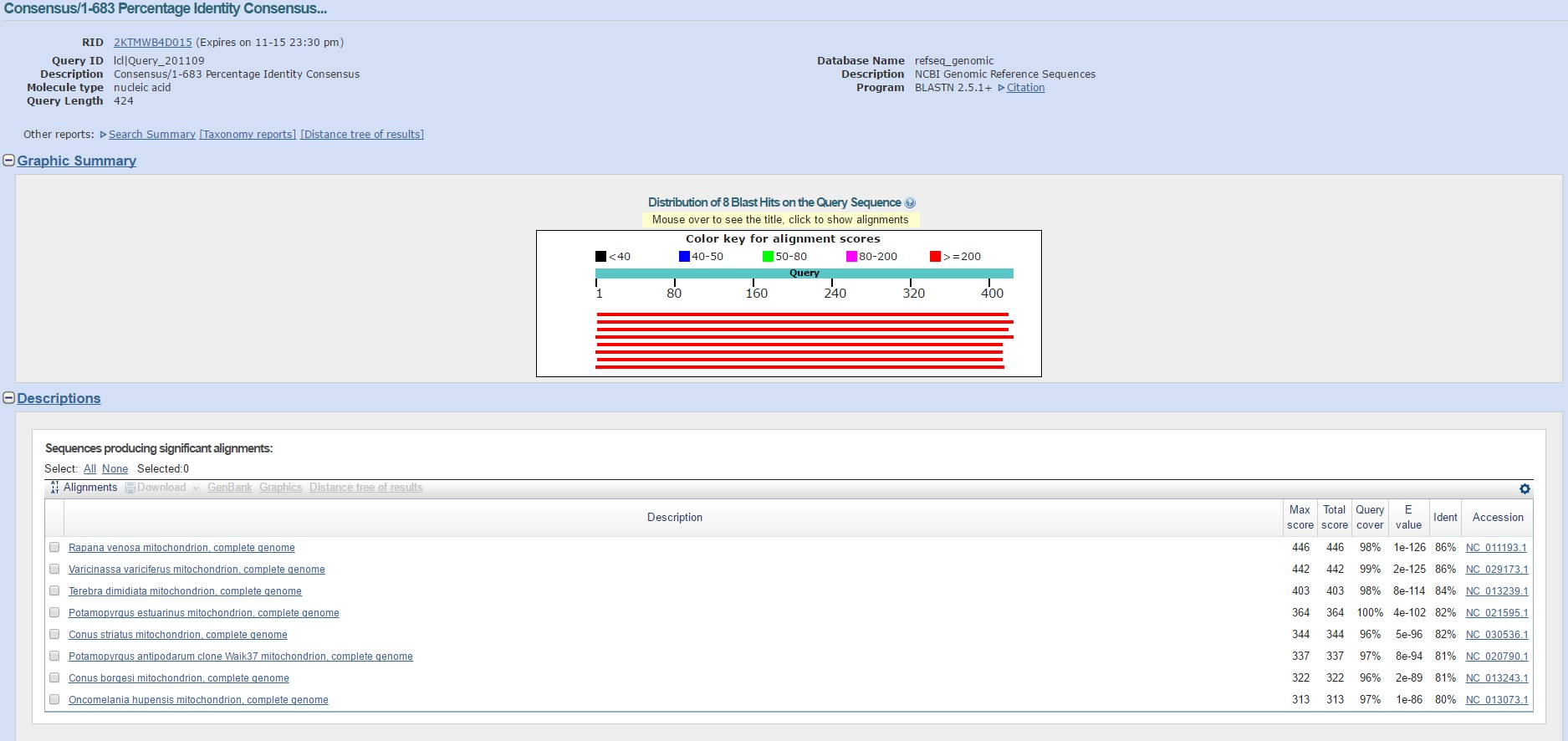
Результаты выдачи blastn
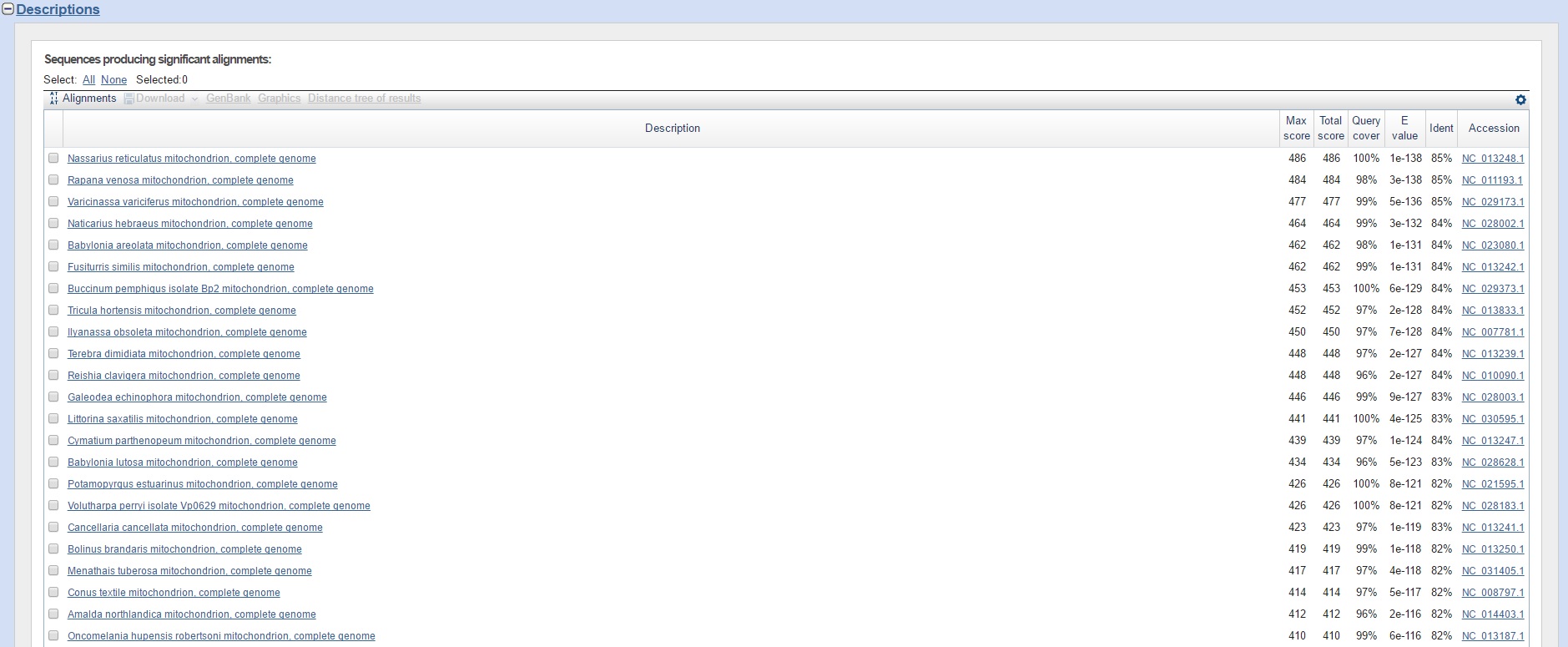
Результаты выдачи discontiguous megablast
| Сравнение алгоритмов |
|---|
| Алгоритм | Число находок | Score лучшей находки | Score худшей находки |
E-value лучшей находки | E-value худшей находки | Ident лучшей находки |
Ident худшей находки | Query cover лучшей находки | Query cover худшей находки |
| megablast | 8 | 446 | 313 | 1e-126 | 1e-86 | 86% | 80% | 98% |
97% |
| discontiguous megablast | 43 | 486 | 311 | 1e-138 | 4e-86 | 76% | 100% | 100% |
99% |
| blastn | 43 | 486 | 311 | 1e-138 | 4e-86 | 85% | 76% | 100% | 99% |
Как можно заметить, не по всем трем алгоритмам совпадают лучшие находки. Например, лучшая находка Nassarius reticulatus mitochondrion, complete genome по алгоритмам blastn и discontiguous megablast не присутствует вообще в списке
находок megablast. Это связано с параметром минимальной длины слова(28): в соответствующем выравнивании найденной и исходной последовательностей слов такой длины без несовпадений и гэпов нет. Вторая же находка
(Rapana venosa mitochondrion, complete genome) у blastn и discontiguous megablast присутсвует также и в списке, полученном с помощью megablast.
Можно привести достаточно много примеров находок, которые нашлись при работе blastn и discontiguous megablast, но их не в списке megablast. Вот одна из них: Eualetes tulipa mitochondrion, complete genome.
Проведенное сравнение позволяет сделать вывод, что алгоритмы blastn и discontiguous megablast при использованных параметрах работают примерно одинаково и выдают не сильно отичающееся число находок сходного качества(в моем случае,
списки находок идентичны). Megablast работает гораздо строже, отсеивая большее количество находок и, следовательно, выдавая последовательности, лишь наиболее близкие к исходной. У megablast относительно высокие требования к E-value,
а Query cover не может быть ниже 28 нуклеотидов, поэтому он может не находить короткие последовательности.
Проверка наличия гомологов белков
В задании необходимо было проверить наличие гомологов трех белков в геноме организма X5 (Amoboaphelidium protococarum) c помощью BLAST+. Была создана локальная база данных с помощью команды makeblastdb и ее опций
-in и -dbtype. Для проверки гомологии были выбраны следующие белки: HSP7C_HUMAN, TERT_HUMAN, CISY_HUMAN. После этого, с помощью алгоритма tblastn, был запущен поиск гомологов по локальной базе данных. Результаты работы
программы и краткие аннотации к белкам представлены ниже.
Белок HSP7C_HUMAN
HSP7C_HUMAN - консервативный шаперон HSP70, белок теплового шока. Имеется у большинства организмов из всех царств, является репрессором активации транскрипции.
Связывая бактериальные ЛПС (липополисахариды), играет роль посредника в процессах ЛПС-индуцированного воспаления, включая секрецию моноцитами TNF (фактора некроза
опухоли).[3]
Результаты tblastn по базе данных X5.fasta для данного белка: hsp7c.out. Всего находок по локальной базе данных - 16.
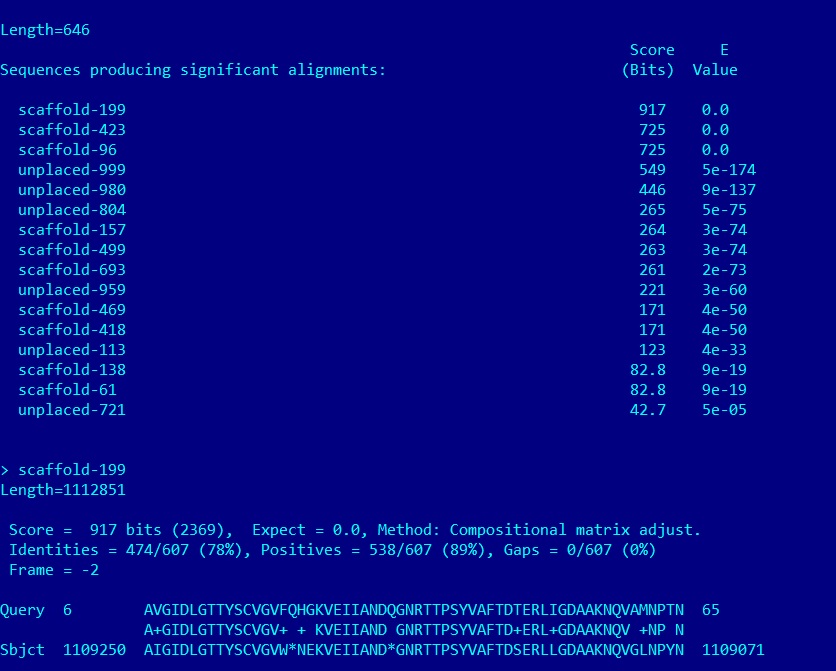 Параметры лучшей находки:
> scaffold-199
Length=1112851, Score = 917 bits (2369), Expect = 0.0, Identities = 474/607 (78%), Positives = 538/607 (89%), Gaps = 0/607 (0%), Frame = -2;
У данной находки хороший E-value, достаточно высокие значения Positives и Identities. Параметры сходства я считаю удовлетворительными, чтобы назвать ее гомологом исследуемого белка, вероятно выполняющим ту же функцию.
Параметры лучшей находки:
> scaffold-199
Length=1112851, Score = 917 bits (2369), Expect = 0.0, Identities = 474/607 (78%), Positives = 538/607 (89%), Gaps = 0/607 (0%), Frame = -2;
У данной находки хороший E-value, достаточно высокие значения Positives и Identities. Параметры сходства я считаю удовлетворительными, чтобы назвать ее гомологом исследуемого белка, вероятно выполняющим ту же функцию.
Белок TERT_HUMAN
TERT_HUMAN - теломераза, восстанавливающая длину теломер(концевых участков хромосом) при репликации. Имеется у большинства, но не у всех эукариот. Активна в раковых клетках и их предшественниках, в соматических же
не активна или проявляет очень низкую активность. Играет важную роль в процессах старения и развития апоптоза.[4]
Результаты tblastn по базе данных X5.fasta для данного белка: tert.out. Всего находок по локальной базе данных - 3.
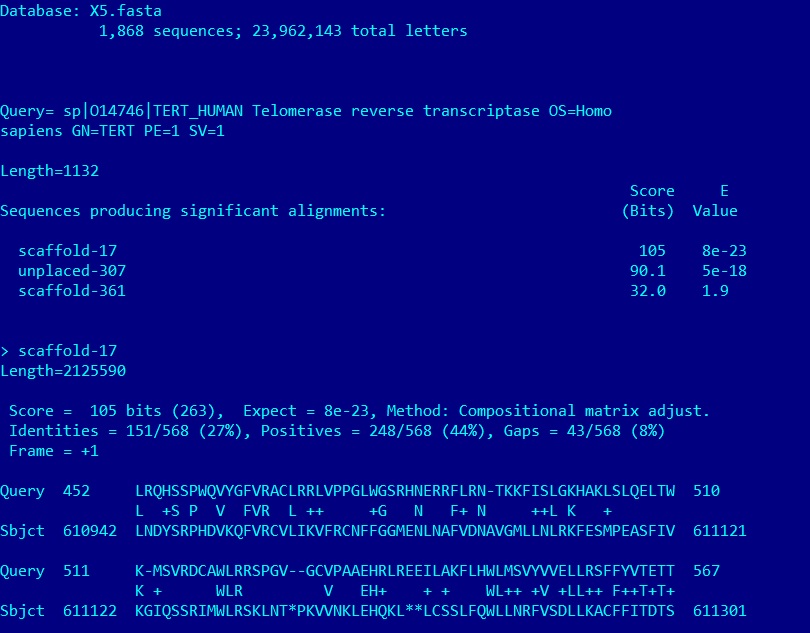 Параметры лучшей находки:
> scaffold-17
Length=2125590, Score = 105 bits (263), Expect = 8e-23, Identities = 151/568 (27%), Positives = 248/568 (44%), Gaps = 43/568 (8%), Frame = +1;
Результат является условно положительным: tblastn выдал три находки, однако даже у самой лучшей из них параметры сходства слишком низкие, чтобы утверждать сохранение функций белка.
Параметры лучшей находки:
> scaffold-17
Length=2125590, Score = 105 bits (263), Expect = 8e-23, Identities = 151/568 (27%), Positives = 248/568 (44%), Gaps = 43/568 (8%), Frame = +1;
Результат является условно положительным: tblastn выдал три находки, однако даже у самой лучшей из них параметры сходства слишком низкие, чтобы утверждать сохранение функций белка.
Белок CISY_HUMAN
CISY_HUMAN - цитратсинтаза митохондрий. Участвует в цикле трикарбоновых кислот(цикл Кребса): проявляет каталитическую активность в реакции Acetyl-CoA + H2O + oxaloacetate = citrate + CoA.[5]
Результаты tblastn по базе данных X5.fasta для данного белка: cisy.out. Всего находок по локальной базе данных - 4.
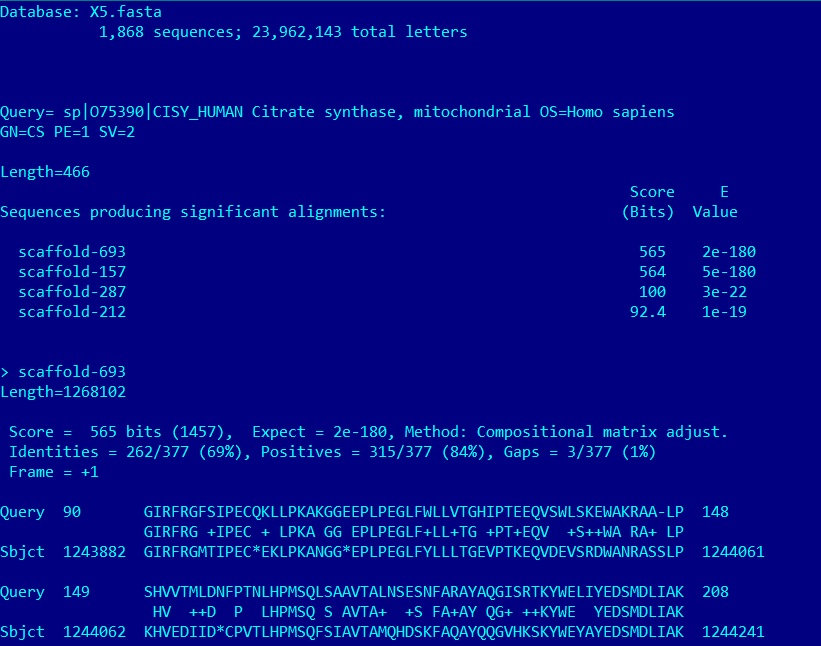 Параметры лучшей находки:
> scaffold-693
Length=1268102, Score = 565 bits (1457), Expect = 2e-180, Identities = 262/377 (69%), Positives = 315/377 (84%), Gaps = 3/377 (1%), Frame = +1;
В силу параметров лучшей находки ее можно назвать гомологом CISY_HUMAN с сохранением функции исследуемого белка.
Параметры лучшей находки:
> scaffold-693
Length=1268102, Score = 565 bits (1457), Expect = 2e-180, Identities = 262/377 (69%), Positives = 315/377 (84%), Gaps = 3/377 (1%), Frame = +1;
В силу параметров лучшей находки ее можно назвать гомологом CISY_HUMAN с сохранением функции исследуемого белка.
Поиск гена белка, закодированного в одном контиге ''Amoboaphelidium''
*Поиск проводился против базы данных Reference genomic sequences из-за реорганизации сайта и невозможности ограничить таксономически поиск по базе Nucleotide collection.
Для поиска был выбран скэффолд scaffold-698. Его длина составляет 23460 п.н., т.е. на нем вполне может поместиться ген. Информация о длинах скэффолдов была получена с помощью команды infoseq X5.fasta -only -name -length.
Командой seqret X5.fasta: scaffold-698 -out scaffold-698.fasta выбранный скэффолд был извлечен в отдельный файл (scaffold-698.fasta).
Поиск проводился против базы данных Reference genomic sequences. Перед выбором таксономического
ограничения я проверила количество отсеквенированных и внесенных в базу последовательностей, принадлежих близким таксонам (например, семейству). Таких последовательностей оказалось совсем мало и полногеномных среди них,
разумеется, не было. По этой причине я выставила ограничение по таксону Fungi/Metazoa group (taxid:33154).
Поиск был произведен с помощью алгоритма blastx, т.к. для поиска гомологии между кодирующими последовательностями полезно транслировать поданную на вход нуклеотидную последовательность в белки. Результат на выходе получается
более достоверный и обоснованный.
Лучшие результаты blastx
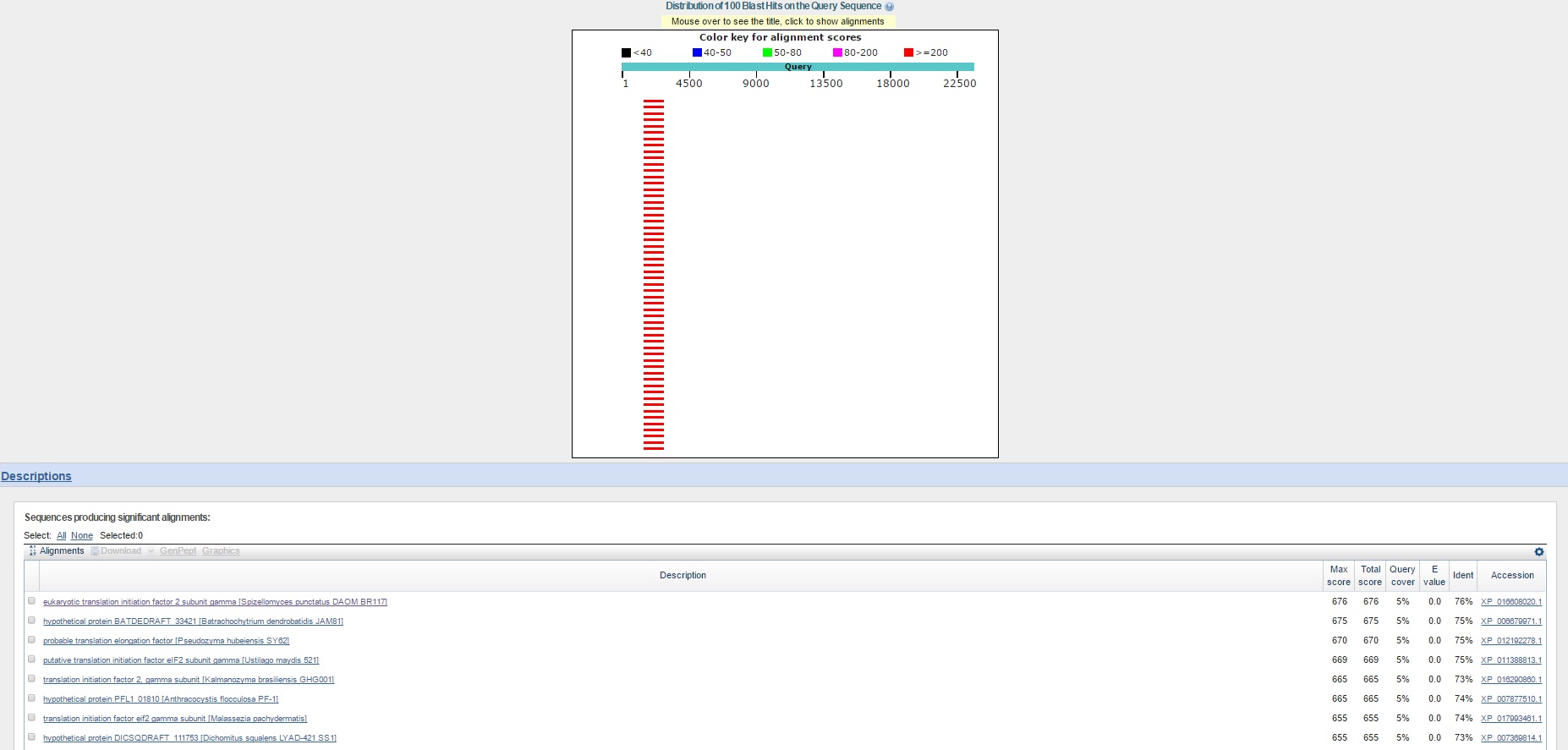
Как можно видеть, scaffold-698 хорошо выравнивается с участками последовательностей других представителей таксона. Показатели веса находок: min E-value = 0.0, max Ident = 76%, Query cover = 5%, Max score = 676.
Исходя из показателей веса выравнивания можно утверждать, что такие участки гомологичны. Ген, находящийся в этом скэффолде, кодирует гамма-субъединицу белка-фактора инициации трансляции - процесса узнавания
рибосомой старт-кодона (AUG-кодона), кодирующего метионин и привлечения инициаторной аминоацил-тРНК.[6]
Привожу ссылку на первую находку eukaryotic translation initiation factor 2 subunit gamma [Spizellomyces punctatus DAOM BR117].
Источники:
[1] Цитохром С-оксидаза
[2]Lacuna vincta
[3]HSP7C_HUMAN
[4]TERT_HUMAN
[5]CISY_HUMAN
[6]Трансляция Wiki
©
Avdiunina Polina, 2015