EMBOSS. Выравнивание геномов.
Таксономия и функция последовательности
1.(seqret) Несколько файлов в формате fasta собрать в единый файл.
При выполнении задания были использованы белковые последовательности hsp7c_human.fasta, cisy_human.fasta и tert_human.fasta из практикума 8.
Команда:seqret "*human.fasta" human_all.fasta
Итоговый файл:human_all.fasta
2.(seqretsplit) Один файл в формате fasta с несколькими последовательностями разделить на отдельные fasta файлы.
При выполнении задания были использованы белковые последовательности из файла human_all.fasta(см. предыдущее задание).
Команда:seqret human_all.fasta
Итоговые файлы:hsp7c_human.fasta, cisy_human.fasta и tert_human.fasta
3.(seqret) Из файла с хромосомой в формате .gb вырезать три кодирующих последовательности по указанным координатам "от", "до", "ориентация" и сохранить в одном fasta файле.
Для выполнения задания я использовала полногеномную последовательность Escherichia coli O157:H7 str. Sakai(BA000007.2). Были найдены три кодирующие области,чьи координаты записаны в файл list_2.txt,
который затем подавался на вход команде.
Команда:seqret @list.txt fasta:final_3.fasta
Итоговый файл:final_3.fasta
4.(transeq) Транслировать кодирующие последовательности, лежащие в одном fasta файле, в аминокислотные, используя указанную таблицу генетического кода. Результат - в одном fasta файле.
Полученные в предыдущем задании кодирующие последовательности были транслированы в последовательности аминокислот, используя стандартную таблицу генетического кода (параметр -table 0). Количество
последовательностей в стартовом и итоговом файле были одинаковы.
Команда:transeq -table 0 final_3.fasta protein_1.fasta
Итоговые файлы:protein_1.fasta
5.(transeq) Транслировать данную нуклеотидную последовательность в шести рамках.
При добавлении к команде transeq опции -frame 6, нуклеотидная последовательность транслируется в аминокислотную в 6-ти рамках. В выходном файле оказалось 18 последовательностей (6*3).
Команда:transeq -frame 6 finak_3.fasta protein_6.fasta
Итоговые файлы:protein_6.fasta
6.(seqret) Перевести выравнивание из fasta формата в формат .msf.
Файл, поданный на вход - seq_test.fasta. Файл, полученный на выходе - seq_test.msf, в котором содержится дополнительная информация о последовательностях. В процессе выполнения команды(см. ниже)
программа запросила имя входного файла.
Команда:seqret -outseq msf::seq_test.msf
Итоговые файлы:seq_test.msf
7.(infoalign) Выдать в выходной поток число совпадающих букв между второй последовательностью выравнивания и всеми остальными (на выходе только имя последовательности и число).
Команда infoalign выдает различную информацию о последовательностях во входном множественном выравнивании (USA, имя, длину, количество гэпов, совпадений и пр.) в сравнении с выбранной(эталонной)
последовательностью. По умолчанию эталонной последовательностью является консенсусная последовательность, которую вычисляет программа, но ее можно задать и вручную по имени или порядковому номеру в файле.
В задании необходимо было провести сравнение со второй последовательностью, поэтому была использована опция -refseq 2. Чтобы получить на выходе только имя и информацию о количестве совпадений были
использованы опции -only -name -idcount. На вход был подан файл seq_infoalign.fasta.
Команда:infoalign seq_infoalign.fasta -refseq 2 -only -name -idcount
Итоговые файлы:seq_output_1.txt
8.(featcopy) Перевести аннотации особенностей в записи формата .gp в табличный формат .gff.
Команда featcopy читает таблицы особенностей и переводит их в любой из поддерживаемых форматов. Исходный файл - sequence.gp.
Команда:featcopy sequence.gp sequence_1.gff
Итоговые файлы:sequence_1.gff
9.(extractfeat) Из данного файла с хромосомой в формате .gb получить fasta файл с кодирующими последовательностями; (*) добавить в описание каждой последовательности функцию белка (из поля product).
Команда extractfeature предназначена для извлечения участков последовательности, аннотированных какой-дибо особенностью. С помощью опции -type можно указать конкретное название поля особенности.
Участки с одной и той же особенностью могут быть извлечены как отдельные последовательности или сцеплены вместе, если используется опция -join. Необходимо было получить файл с кодирующими последовательностями,
поэтому команда была запущена с опцией -type CDS. Чтобы добавить в описание функцию белка была также использована опция -describe. Исходный файл - sequence.gb.
Команда:extractfeat sequence.gb -type CDS -describe product extr.fasta
Итоговые файлы:extr.fasta
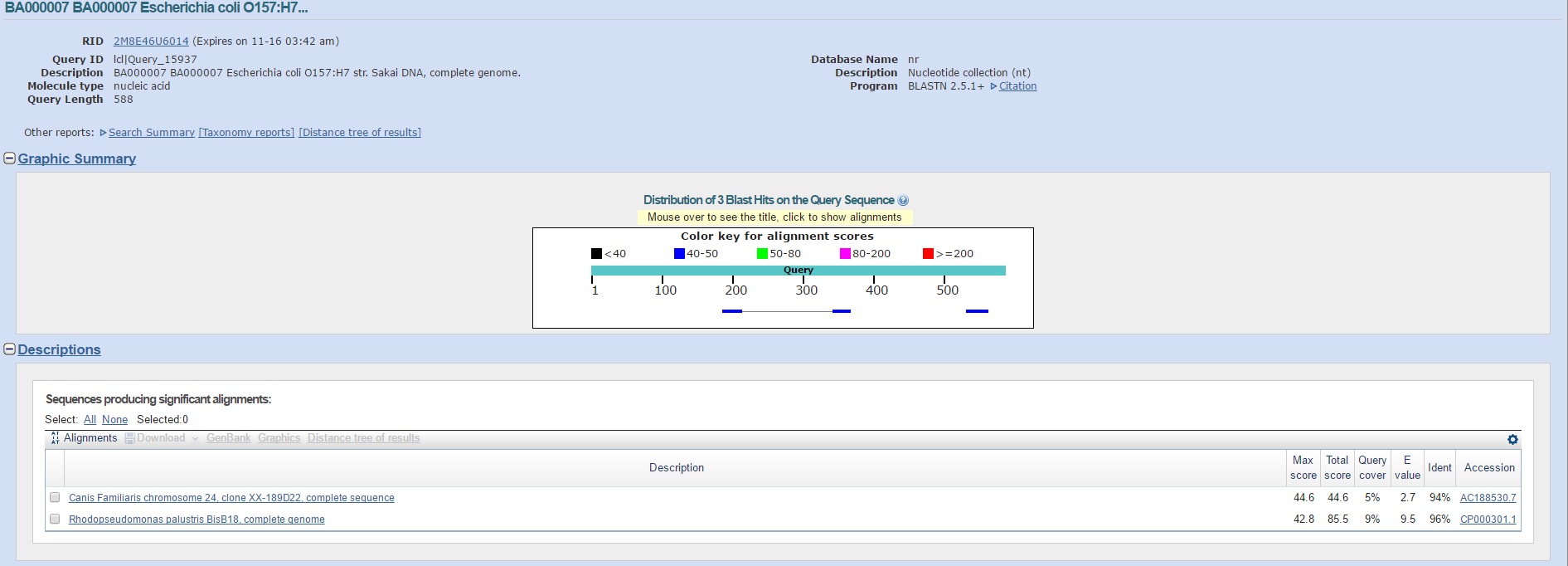
10.(shuffle) Перемешать буквы в данной нуклеотидной последовательности; (*) проверить с помощью blastn сколько "достоверных" находок (с E-value < 0.1) найдется в нуклеотидном банке данных
(запустите с порогом E = 10 - по умолчанию).
Команда shuffle читает одну или несколько последовательностей и случайным образом перемешивает их. Число перемешиваний может быть задано. Для работы был взят файл snuf_1.fasta.
Команда:shuffleseq snuf.fasta -outseq snuf_1.fasta
Итоговые файлы:snuf_1.fasta
Для выходного файла был запущен blastn с параметрами по умолчанию. "Достоверных" находок (с E-value < 0.1) не нашлось ни одной. Выдачу blastn можно увидеть на рисунке.
 11.(cusp) Найдите частоты кодонов в данных кодирующих последовательностях.
Команда cusp высчитывает таблицу использования кодонов для одной или более нуклеотидных последовательностей. В данной таблице для каждого кодона указано: последовательность кодона, закодированная
аминокислота, частота данного кодона по отношению к остальным, кодирующим ту же аминокислоту (fraction), частота кодона в данной последовательности (frequency), число кодонов данного типа в последовательности.
Для работы был взят файл snuf_1.fasta.
Команда:cusp snuf.fasta snuf.cusp
Итоговые файлы:snuf.cusp
12.(compseq) Найдите частоты динуклеотидов в данной нуклеотидной последовательности и сравните их с ожидаемыми.
Команда compseq производит подсчет состава слов указанной длины во входной последовательности. В выходной файл записывается само слово, сколько раз оно встречается, его наблюдаемая частота встречаемости (Obs Frequency),
предполагаемая частота (Exp Frequency), а также соотношение этих частот (Obs/Exp Frequency). Чтобы найти наблюдаемые и ожидаемые частоты динуклеотидов необходимо запустить данную команду для слов длины 2 (опция -word 2).
Для более точного подсчета ожидаемых частот можно использовать опцию -calcfreq, при активировании которой данный подсчет ведется на основании частот встречаемости отдельных оснований в данной последовательности.
Исходный файл - snuf_1.fasta.
Команда:compseq -word 2 -calcfreq snuf.fasta snuf.compseq
Итоговые файлы:snuf.compseq
13.(tranalign) Выровняйте кодирующие последовательности соответственно выравниванию белков - их продуктов.
Команда tranalign принимает на вход набор из невыровненных нуклеотидных последовательностей и соответсвующий им набор выровненных транслированных с них белковых последовательностей. В выходной файл записывается выравнивание
нуклеотидных последовательностей. Каждая нуклеотидная последовательность транслируется во всех трех прямых рамках по указанному генетическому коду и трансляции сравниваются с соответсвующими во входном выравнивании. Важно, чтобы
соответствующие друг друг последовательности во входных файлах располагались в одном порядке. Для работы были взяты следующие файлы: - невыровненные нуклеотидные последовательности, - выровненные белковые последовательности.
Команда:
Итоговые файлы:
11.(cusp) Найдите частоты кодонов в данных кодирующих последовательностях.
Команда cusp высчитывает таблицу использования кодонов для одной или более нуклеотидных последовательностей. В данной таблице для каждого кодона указано: последовательность кодона, закодированная
аминокислота, частота данного кодона по отношению к остальным, кодирующим ту же аминокислоту (fraction), частота кодона в данной последовательности (frequency), число кодонов данного типа в последовательности.
Для работы был взят файл snuf_1.fasta.
Команда:cusp snuf.fasta snuf.cusp
Итоговые файлы:snuf.cusp
12.(compseq) Найдите частоты динуклеотидов в данной нуклеотидной последовательности и сравните их с ожидаемыми.
Команда compseq производит подсчет состава слов указанной длины во входной последовательности. В выходной файл записывается само слово, сколько раз оно встречается, его наблюдаемая частота встречаемости (Obs Frequency),
предполагаемая частота (Exp Frequency), а также соотношение этих частот (Obs/Exp Frequency). Чтобы найти наблюдаемые и ожидаемые частоты динуклеотидов необходимо запустить данную команду для слов длины 2 (опция -word 2).
Для более точного подсчета ожидаемых частот можно использовать опцию -calcfreq, при активировании которой данный подсчет ведется на основании частот встречаемости отдельных оснований в данной последовательности.
Исходный файл - snuf_1.fasta.
Команда:compseq -word 2 -calcfreq snuf.fasta snuf.compseq
Итоговые файлы:snuf.compseq
13.(tranalign) Выровняйте кодирующие последовательности соответственно выравниванию белков - их продуктов.
Команда tranalign принимает на вход набор из невыровненных нуклеотидных последовательностей и соответсвующий им набор выровненных транслированных с них белковых последовательностей. В выходной файл записывается выравнивание
нуклеотидных последовательностей. Каждая нуклеотидная последовательность транслируется во всех трех прямых рамках по указанному генетическому коду и трансляции сравниваются с соответсвующими во входном выравнивании. Важно, чтобы
соответствующие друг друг последовательности во входных файлах располагались в одном порядке. Для работы были взяты следующие файлы: - невыровненные нуклеотидные последовательности, - выровненные белковые последовательности.
Команда:
Итоговые файлы:
Сравнение геномов
Построение карты локального сходства
Для выполнения задания были взяты бактерии Yersinia pestis Nepal516 и Yersinia pestis Antiqua. Это патогенные бактерии, вызывающие чуму.
С помощью blast2seq было построено выравнивание и получена карта локального сходства для этих двух штаммов.
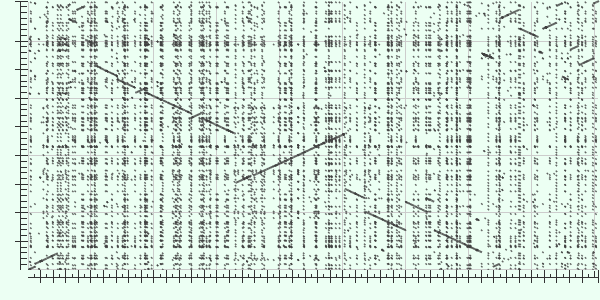 |
| Карта локального сходства Yersinia pestis Nepal516 (OX) и Yersinia pestis Antiqua (OY) |
|---|
Анализ полученнной карты
Данная карта (dot matrix view) показывает участки сходства на основании результатов работы BLAST. Выравнивания показаны
в виде непрерывных линий. Совпадения прямой цепи (plus strand) отображены линиями, идущими из левого нижнего в правый
верхний угол, совпадения комплементарной цепи (minus strand) - из левого верхнего в правый нижний. Количество линий
соответсвует количеству найденных BLAST выравниваний.
Последовательность на оси OY (Query) имеет длину 4702289, полседовательность на оси OX (Reference) - 4534590. Минимальная цена деления на осях составляет
50000.
Cходство (Identity %) между гомологичными участками в данном выравнивании - 99% (было найдено
как среднее сходство по нескольким наиболее длинным выравниваниям).
| Синтеничные области |
|---|
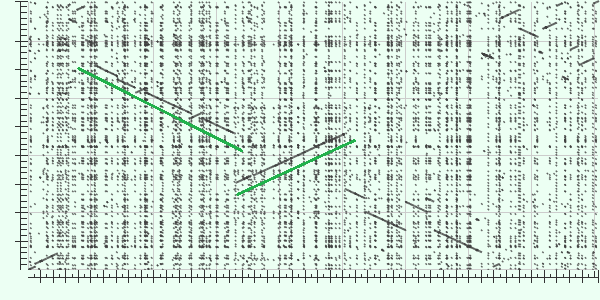 | Синтеничные области - участки геномов, состоящие
из ортологичных областей с сохранением их порядка на хромосоме для сравниваемых геномов. Наиболее крупные
синтеничные области для геномов Yersinia pestis Nepal516 (OX) и Yersinia pestis Antiqua (OY) подчеркнуты зеленым. |
| Наиболее крупные эволюционные события |
|---|
 | Рис. 1
Красным выделены инверсии. |
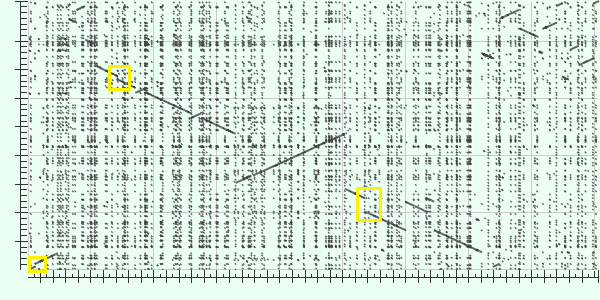 | Рис. 2
Желтым выделены вставки в Query |
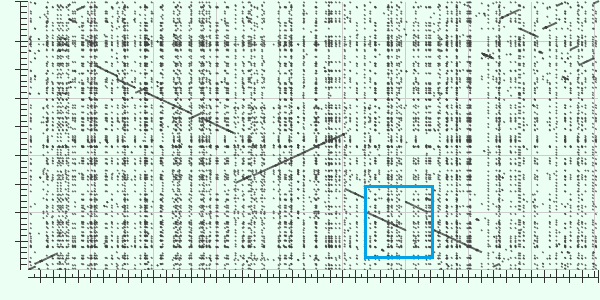 | Рис. 3
В выделенной голубым области вероятно
произошла транслокация. Наблюдается перестановка порядка участков - AB в одном геноме, BA - в другом. |
Источники:
[1] Цитохром С-оксидаза
[2]Lacuna vincta
©
Avdiunina Polina, 2015