 Программа в той же папке выдала мне файл chr2_fastqc.html, содержащий визуализированный отчёт о работе, и архив chr2_fastqc.zip с каринками и текстовыми файлами.
Программа в той же папке выдала мне файл chr2_fastqc.html, содержащий визуализированный отчёт о работе, и архив chr2_fastqc.zip с каринками и текстовыми файлами.
Для выполнения задания был выдан файл с одноконцевыми чтениями экзома 2 хромосомы человека chr2.fastq, и файл с последовательностью этой хромосомы из генома со сборкой версии hg19 chr2.fasta.
Все действия выполнялись в специально созданной директории /nfs/srv/databases/ngs/p.avdiunina
Сперва был выполнен анализ качества чтений с помощью программы FastQC(использовалась версия, установленная на сервере kodomo). В командную строку была введена команда:
Программа в той же папке выдала мне файл chr2_fastqc.html, содержащий визуализированный отчёт о работе, и архив chr2_fastqc.zip с каринками и текстовыми файлами.
Далее производилась очистка чтений при помощи программы Trimmomatic, которая также была запущена с сервера kodomo следующей командой:
 Первая опция отрезает с конца каждого чтения нуклеотиды с качеством ниже 20, вторая удаляет риды короче 50 нуклеотидов. До чистки было 10410 чтений, после - осталось 10191 (удалено 219 чтений).
Затем снова была запущена программа FastQC, результатом стали архив out_chr2_fastqc.zip и файл out_chr2_fastqc.html
С помощью команды unzip *.zip были разархивированы оба архива. Теперь выполним сравнение чтений до и после очистки.
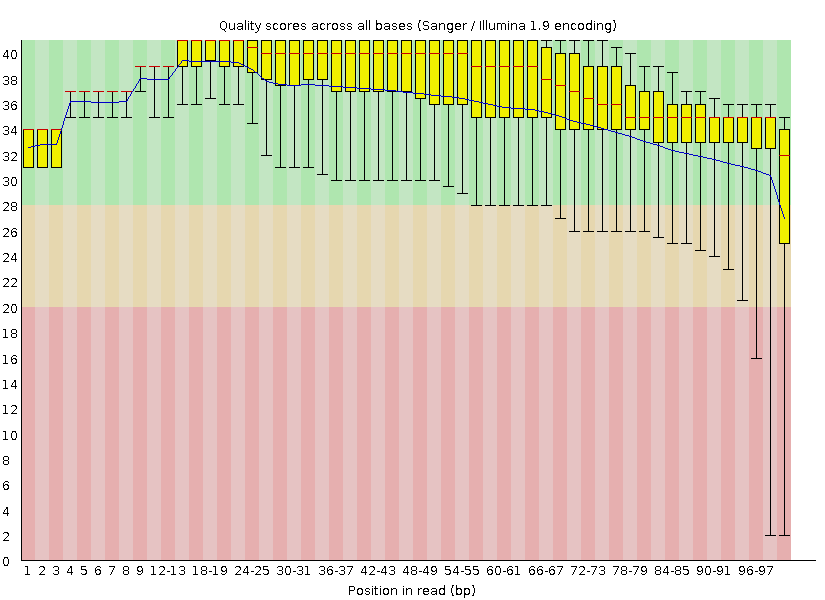
На рис. 1 представлен график качества чтений, сделанный программой FastQC до чистки. Синей линией на графике обозначено среднее качество чтений, центральными красными линиями обозначена медиана, желтыми
прямоугольниками - интерквартальный размах (разница между верхней и нижней квартилями, диапазон значений качества, при котором качество 25% чтений на данной позиции выше нижней границы, а 75% - не выше верхней).
Поле графика разделено на 3 полосы зеленого, желтого и красного цветов, попадание в которые вышеперечисленных элементов графика позволяет судить о качестве чтений.
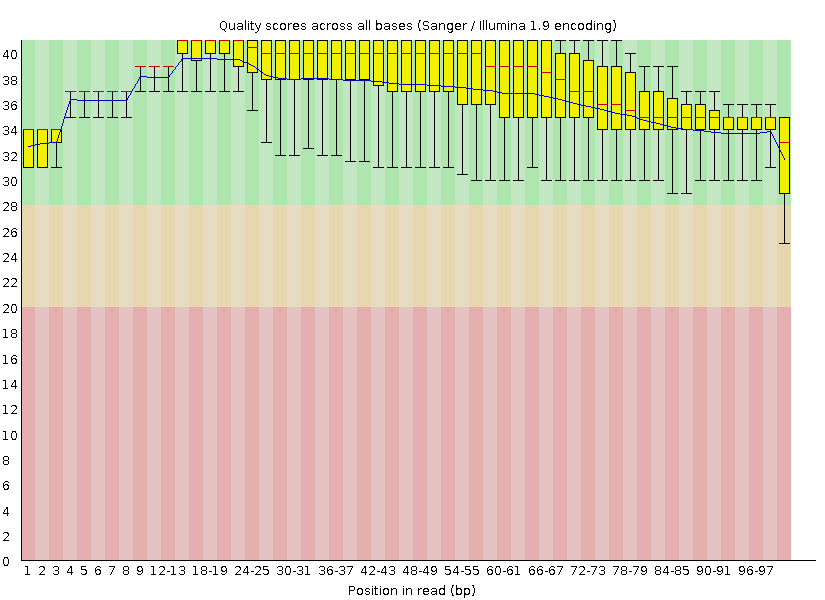
На рис. 2 представлен график качества чтений, сделанный программой FastQC после очистки. Теперь все позиции, кроме последней, расположены в зеленой области (последняя - в желтой), из чего можно заключить,
что чистка прошла успешно. Еще один результат работы программы Trimmomatic состоял в удалении чтений с длиной меньше 50 нуклеотидов, теперь длины чтений колеблются от 50 до 100 нуклеотидов (раньше: от 40 до 100).
Первая опция отрезает с конца каждого чтения нуклеотиды с качеством ниже 20, вторая удаляет риды короче 50 нуклеотидов. До чистки было 10410 чтений, после - осталось 10191 (удалено 219 чтений).
Затем снова была запущена программа FastQC, результатом стали архив out_chr2_fastqc.zip и файл out_chr2_fastqc.html
С помощью команды unzip *.zip были разархивированы оба архива. Теперь выполним сравнение чтений до и после очистки.
На рис. 1 представлен график качества чтений, сделанный программой FastQC до чистки. Синей линией на графике обозначено среднее качество чтений, центральными красными линиями обозначена медиана, желтыми
прямоугольниками - интерквартальный размах (разница между верхней и нижней квартилями, диапазон значений качества, при котором качество 25% чтений на данной позиции выше нижней границы, а 75% - не выше верхней).
Поле графика разделено на 3 полосы зеленого, желтого и красного цветов, попадание в которые вышеперечисленных элементов графика позволяет судить о качестве чтений.
На рис. 2 представлен график качества чтений, сделанный программой FastQC после очистки. Теперь все позиции, кроме последней, расположены в зеленой области (последняя - в желтой), из чего можно заключить,
что чистка прошла успешно. Еще один результат работы программы Trimmomatic состоял в удалении чтений с длиной меньше 50 нуклеотидов, теперь длины чтений колеблются от 50 до 100 нуклеотидов (раньше: от 40 до 100).
 |
 |
|---|---|
| Рис. 1. "Per base sequence quality", график качества чтений, сделанный программой FastQC до чистки | Рис. 2. "Per base sequence quality", график качества чтений, сделанный программой FastQC после чистки |
Чтения были откартированы с помощью программы BWA - Burrows-Wheeler Alignment Tool. Использованные команды указаны в таблице.
| Команда | Назначение | Результат |
|---|---|---|
| bwa index chr2.fasta | Индексирование референсной последовательности | Индексированный файл chr2.fasta |
| bwa mem chr2.fasta out_chr2.fastq > first_align.sam | Выравнивание очищенных чтений с референсной последовательностью |
Файл first_align.sam, содержащий выравнивание в формате SAM (Sequence Alignment/Map) |
Для анализа полученного выравнивания применялась программа Samtools, предназначенная для обработки файлов в формате SAM. Использованные команды указаны в таблице.
| Команда | Назначение | Результат |
|---|---|---|
| samtools view first_align.sam -bo first_align.bam | Перевод выравнивания в бинарный формат .bam (опция -b меняет формат выходного файла с установленного по умолчанию, -o обозначат имя выходного файла). |
first_align.bam |
| samtools sort first_align.bam -T nexact.txt -o align_sort.bam | Сортировка выравнивания чтений с референсом по координате в референсе начала чтения (опция -T позволяет записывать временные файлы в файл smth.txt, а не в stdout). |
align_sort.bam |
| samtools index align_sort.bam | Индексирование отсортированного выравнивания | Индексированный align_sort.bam |
| samtools idxstats align_sort.bam > finish.txt | Выяснение числа чтений, откартированных на геном | finish.txt |
| Команда | Назначение | Результат |
|---|---|---|
| samtools mpileup -uf chr2.fasta align_sort.bam > snp.bcf |
Создание файла с полиморфизмами в формате .bcf |
snp.bcf |
| bcftools call -cv snp.bcf -o snp.vcf | Определение отличий между референсом и чтениями |
snp.vcf |
| Координата | Тип полиморфизма |
Было в референсе |
Найдено в чтениях |
Глубина покрытия данного места |
Качество прочтения на участке |
|---|---|---|---|---|---|
| 55516588 | Замена | G | C | 23 | 184.999 |
| 55523307 | Индель | ACC | AC | 9.04379 | |
| 234160243 | Замена | G | C | 7 | 81.0075 |
Далее необходимо было аннотировать полученные SNP с помощью программы ANNOVAR. Для этого сначала нужно было подготовить входной файл, чтобы программа могла работать с ним. В первую очередь из файла snp.vcf были удалены
все индели, полученный файл - snp_noindel.vcf. Затем был использован скрипт convert2annovar.pl с помощью команды perl /nfs/srv/databases/annovar/convert2annovar.pl -format vcf4 /nfs/srv/databases/ngs/p.avdiunina/snp.vcf >
/nfs/srv/databases/ngs/p.avdiunina/snp.avinput.
Далее были проведены аннотирования по БД, указанным выше. Команды:
Источники:
© Avdiunina Polina, 2015