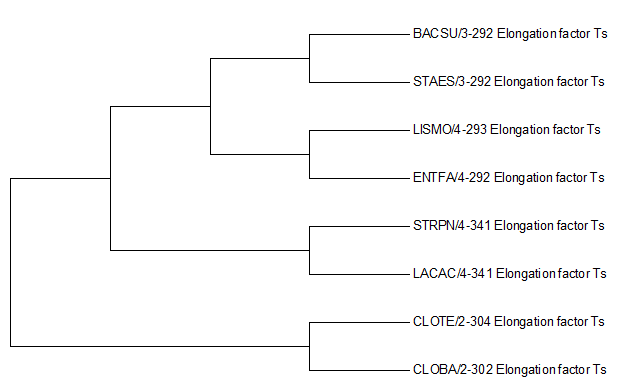
На следующих рисунках показаны блоки(или части блоков), в которых заметна гомологичность всех последовательностей.
Видимо, это активные центры белков. Однако уже в
этих консервативных участках наблюдаются некоторые отклонения,
отраженные на дереве. Например, на первом рисунке видны отличия белков из CLOTE и
CLOBA от остальных: нет гэпов,
в позиции 168 находится алифатическая аминокислота валин(V)(у остальных последовательностей нейтральные серин и
треонин),
в позиции 169 находится глутаминовая кислота(E)(у остальных алифатические аминокислоты в данной позиции),
в позиции 189 у CLOTE и CLOBA глутамин(Q), у
остальных же гистидин(H).
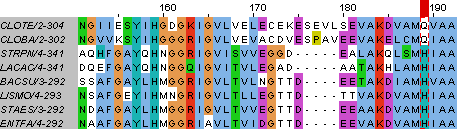
Рис.1 Отличия последовательностей CLOTE и CLOBA от остальных: позиции 168, 169, 189
Похожий пример можно увидеть на рис.2, где
Внутри порядка Lactobacillales(в него входят ENTFA, STRPN, LACAC) можно выделить множество диагностических позиций
JalView предлагает три вида алгоритма Neighbour-Joining: с использованием процента идентичных позиций (% Identity), PAM 250 или BLOSUM62.
Рис.4 Филогенетическое дерево, построенное с использованием алгоритма Neighbour-Joining Using % Identity
Деревья, построенные по алгоритмам Neighbour-Joining BLOSUM62 и Neighbour-Joining PAM250 отличаются между собой расположением последовательности ENTFA относительно
Рис.5 Филогенетическое дерево, построенное с использованием алгоритма Neighbour-Joining Using BLOSUM62
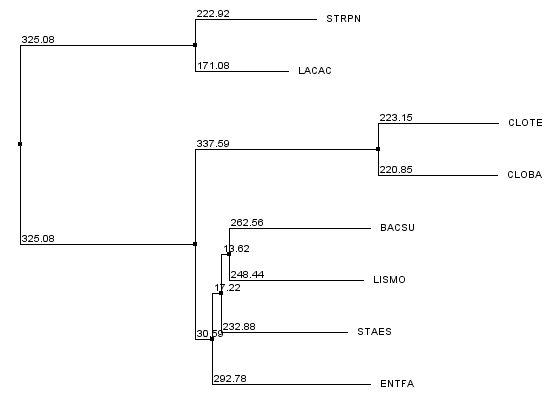
Рис.6 Филогенетическое дерево, построенное с использованием алгоритма Neighbour-Joining Using Pam250
На рисунке 7 представлено дерево, построенное программой MEGA. Оно отличается от предыдущих деревьев и является наиболее приближенным к реальному дереву, полученному
исходя из дерева видов.
Рис.7 Филогенетическое дерево, построенное с использованием алгоритма Neighbour-Joining в программе MEGA.
©
Avdiunina Polina, 2017
в позиции 229 находится ароматический тирозин(T) у CLOTE и CLOBA против алифатических аминокислот в остальних белках.
В целом наблюдается гораздо больше несовпадений в блоках(и между ними) между последовательностями рода Clostridium(в него
входят CLOB1 и CLOTE) и последовательностями класса Bacilli(в него входят ENTFA, LACAC, STRPN, BACSU, LISMO, STAES).
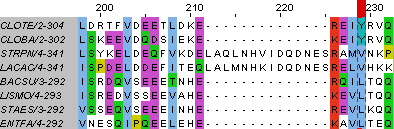
Рис.2 Отличия последовательностей CLOTE и CLOBA от остальных: позиция 229
и целых блоков у последовательностей STRPN и
LACAC(из-за этого по алгоритму Neighbour-Joining с построением матрицы
расстояний через подсчет процента идентичных позиций (% Identity) STRPN и LACAC выделяются
в отдельную кладу).
Примеры на рис.3.
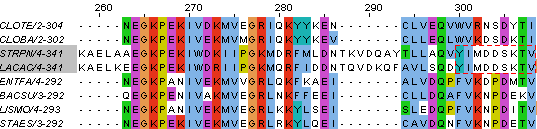
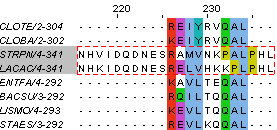
Рис.3 Диагностические позиции и блоки последовательностей STRPN и LACAC
Алгоритм Neighbour-Joining
Также один алгоритм
реализован в программе MEGA.
На рис.4 представлено дерево, построенное по алгоритму Neighbour-Joining % Identity, обзор диагностических позиций см. в задании 1.
По топологии оно
сильно отличается от других деревьев, построенных в Jalview из-за принципиально другого подсчета матрицы расстояний. Различия можно заметить как
во
взаимном расположении бактерий внутри порядков Bacillales и Lactobacillales, так и в расположении этих порядков относительно других таксонов: последовательности
ENTFA, STRPN и LACAC, образующие отдельную кладу на дереве видов, разнесены и по-разному располагаются на каждом из деревьев, например, ENTFA на рис.4
выносится в
отдельную от всего остального дерева ветвь. Последовательности BACSU, LISMO и STAES выносятся в отдельную кладу только на рис.5
и рис.6(Neighbour-Joining BLOSUM62
и Neighbour-Joining PAM250, соответственно), однако их локальная топология различается внутри клады.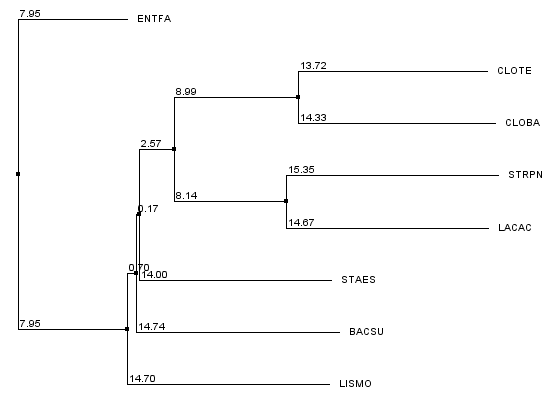
порядка Bacillales и рода Clostridium(на рис.5 ENTFA выделен в отдельную ветвь относительно упомянутых таксонов, а на рис.6 нет), а также локальной топологией
внутри
Bacillales(на рис.5 BASCU и STAES выделены в отдельную кладу, а LISMO находится отдельно, но на рис.6 кладу составляют BASCU и LISMO).
Ни одна из
реализаций этого алгоритма не привела к абсолютно правильному дереву. 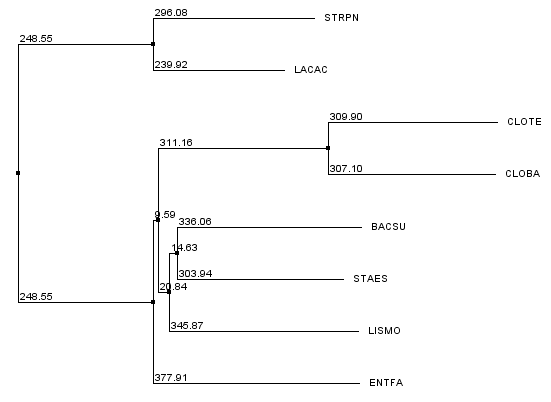
Отличия заключаются во взаимном расположении бактерий внутри класса Bacilli. По дереву видов BACSU, LISMO и STAES составляют отдельную кладу,
выделяющуюся в порядок Bacillales, а ENTFA,
STRPN и LACAC объеденены в кладу, опредляющую порядок Lactobacillales. На дереве, построенном в MEGA, последовательности
ENTFA и LISMO выделены в отдельную кладу, а при укоренении дерева
в ветвь Clostridium(CLOB1, CLOTE), ветвь порядка Lactobacillales(LACAC, STRPN) отходит от ветви класса
Bacilli раньше, чем ветвь порядка Bacillales(BACSU, STAES).