С помощью Muscle было построено выравнивание полученных последовательностей (рисунок 1). Выравнивание в fasta-формате: ali_pr_4.fasta.
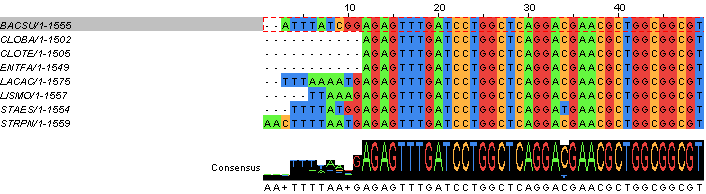
Рис.1 Выравнивание последовательностей 16s рРНК из бактерий. Построено в JalView с помощью Muscle. Раскраска Nucleotide.
С помощью программы MEGA было построено филогенетическое дерево последовательностей по выравниванию. Использованные методы: Neighbor-Joining,
В целом, можно наблюдать существенные различия в качестве построения деревьев по белковым или по нуклеотидным последовательностям: увеличение
В протеомах выбранных бактерий были найдены достоверные гомологи белка CLPX_BASCU. С порогом E-value 0.001 было получено 37 находок (выдача blast),
Гомологичные последовательности называют ортологами, если они произошли в результате видообразования.
Minimum evolution и Maximum likelihood. Все
три метода выдают деревья с одинаковой топологией (рисунок 2). При сравнении с исходным деревом (рисунок 3),
полученным из общего дерева протеобактерий, можно заметить, что у деревьев более
сходная топология, чем у дерева видов и деревьев, построенных по последовательностям
фактора элонгации трансляции Ts (мнемоника EFTS); возросло количество общих ветвей(4 общих ветви). Однако
можно заметить различия во взаимном расположении видов
внутри порядка Bacillales (на рис.5 BASCU и STAES выделены в отдельную кладу, а LISMO находится отдельно, но на рис.3 кладу составляют BASCU
и LISMO)
и отличия в топологии внутри всего класса Bacilli (на рис.3 порядок Lactobacillales выделен в отдельную кладу, тогда как на рис.2 ENTFA, STRPN и LACAC
не соствляют отдельную ветвь).

Рис.2 Филогенетическое дерево, построенное по последовательностям 16s рРНК алгоритмом Neighbor-Joining.
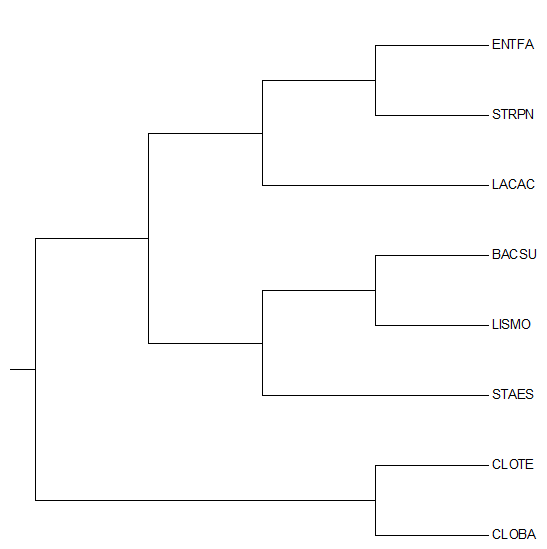
Рис.3 Дерево видов.
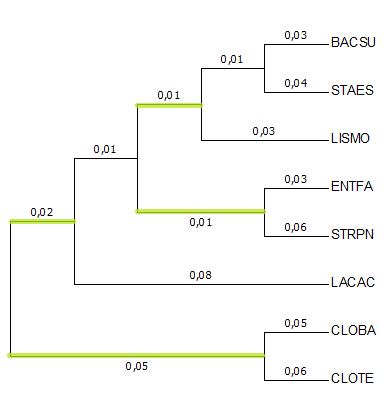
Рис.4 Филогенетическое дерево, построенное по последовательностям 16s рРНК.
Общие в деревом видом ветви выделены цветом.
количества общих ветвей, сходная топология деревьев
при использовании разных алгоритмов и т.д.
Построение и анализ дерева, содержащего паралоги
однако реально
наиболее сходными являются первые 27 находок (с E-value ниже 1e-40).
При помощи fetch sequences последовательности были извлечены в JalView,
и сервисом Muscle было постороено их выравнивание (pr_4_2.fasta). Для удобства в названиях последовательностей были
оставлены только мнемоники.
Выравнивание было открыто в MEGA, и по нему методом Neighbour-joining было построено филогенетическое дерево. (Рис. 3) Считая,
что данное дерево реконструировано верно, можно попытаться найти на нем ортологов и паралогов.

Рис.4 Дерево последовательностей гомологов, построенное методом Neighbour-joining
Паралоги - гомологи, которые произошли в результате дупликации.
В нашем случае два гомологичных белка будем называть ортологами, если они:
а) из разных организмов;
б) разделение их общего
предка на линии, ведущей к ним,
произошло в результате видообразования;
Два гомологичных белка из одного организма будем называть паралогами.