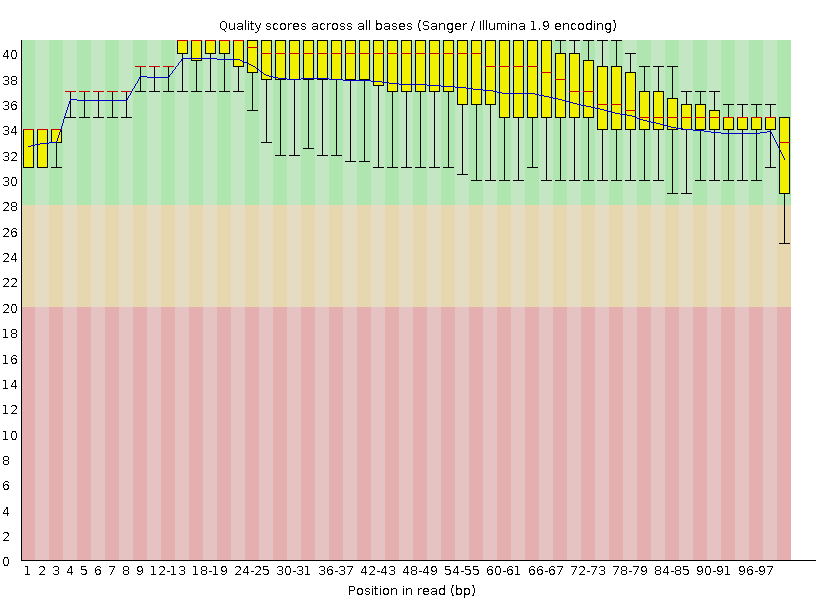
Рис.1 Per Base Sequence Quality, chipseq_chunk54.fastq
Для выполнения задания был выдан файл chipseq_chunk54.fastq с ридами Illumina, полученные в результате сhip-seq эксперимента.
Все действия выполнялись в специально созданной директории /nfs/srv/databases/ngs/p.avdiunina
Сперва был выполнен анализ качества чтений с помощью программы FastQC(использовалась версия, установленная на сервере kodomo).
В командную строку была введена команда: fastqc
chipseq_chunk54.fastq
Программа в той же папке выдала мне файл chipseq_chunk54.html,
содержащий визуализированный отчёт о работе, и архив
chipseq_chunk54.zip с картинками и текстовыми файлами.
Перед очисткой чтений были оценены два графика - распределение качества чтений оснований и длин ридов(см. рисунок 1 и 2).
Длины ридов оказались примерно одинаковы(от 35-37 bp),
качества чтений оснований - от 26 до 38.
Для очистки чтений была использована программой Trimmomatic, убрав риды с качеством ниже 50, остались риды с минимальной длиной 36 пар
нуклеотидов:
java -jar /usr/share/java/trimmomatic.jar SE -phred33 chipseq_chunk54.fastq chipseq_chunk54_out.fastq TRAILING:50 MINLEN:36
Рис.1 Per Base Sequence Quality, chipseq_chunk54.fastq
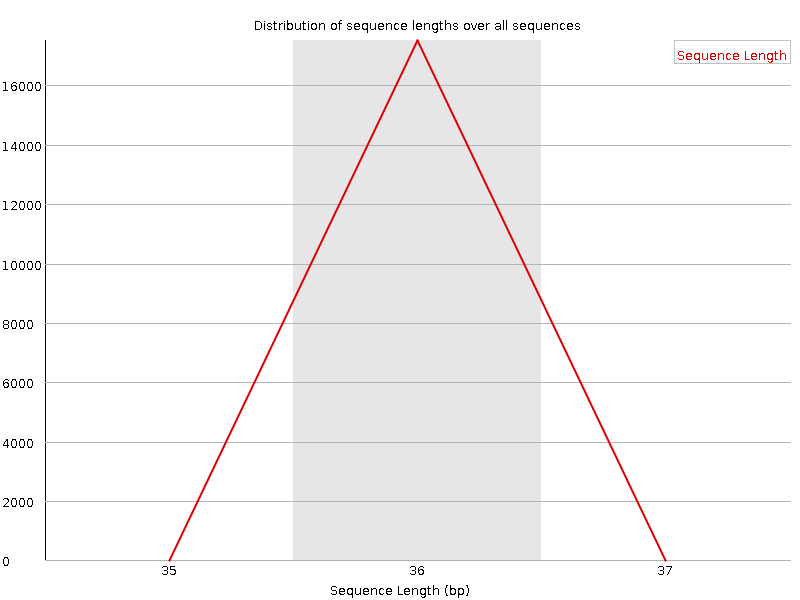
Рис.2 Sequence length distribution, chipseq_chunk54.fastq
Далее последовательности были откартированы на проиндескированный геном человека.
Картирование выполнялось с помощью команды BWA для проиндексированного референса:
bwa mem /srv/databases/ngs/hg19/GRCh37.p13.genome.fa chipseq_chunk54.fastq >
chipseq_chunk54.sam
Далее для анализа картирования было выполнено несколько команд.
Сначала с помощью пакета samtools файл с выравниванием был переведен в бинарный формат .bam:
samtools view chipseq_chunk54.sam -bSo chipseq_chunk54.bam
Получившийся файл chipseq_chunk54.bam был подан на вход другой команде, которая сортирует выравнивание ридов с референсом по координате в референсе
(параметр -T позволяет
записать временные файлы в файл chip_time, т.к. иначе они выводятся в stdout):
samtools sort chipseq_chunk54.bam -T chip_time -o chipseq_chunk54_sorted.bam
Далее полученный файл был проиндексирован:
samtools index chipseq_chunk54_sorted.bam
Затем производился подсчет ридов, которые откартировались на геном:
samtools idxstats chipseq_chunk54_sorted.bam > chip_seq.out
Итоговый файл: chip.out. Таким образом, из 17513 ридов 16290 (93.01%) были откартированы на 4 хромосому, так что предполагаю,
что риды с именно этой хромосомы
были предложены мне для анализа. При этом откартировалось 17032 рида.
Peak calling — компьютерный метод поиска областей генома, на которые откартировалось большое число ридов из данных эксперимента ChiP-Seq.
В случае ТФ эти области соответствуют сайтам
связывания ТФ.
Для поиска пиков использовалась программа MACS (Model-based Analysis of Chip-Seq)
— одна из самых популярных программ для анализа данных ChiP-Seq эксперимента. Для выполнения задания
использовалась следующая команда (параметр
--nomodel введен, так как пиков было слишком мало):
macs2 callpeak -n chipseq_chunk41 -t chipseq_chunk41_sorted.bam --nomodel
При этом было получено 3 файла ( *.narrowPeak, *.xls, *.bed) с информацией о пиках.
Всего было найдено 8 пиков на 4 хромосоме (табл. 2). Визуализация пиков в UCSC Genome Browser
представлена на рис. 3.
Для визуализации в браузер подавался файл chipseq_chunk41_peaks.narrowPeak, в начало которого было дописано
"track type=narrowPeak visibility=3 db=hg19 name="my_peaks" description="Peaks from chunk 54" browser position chr4:74211425-75579621"
| № | Начало | Конец | Длина | -log10(p-value) |
|---|---|---|---|---|
| 1 | 74211426 | 74211710 | 285 | 18.73502 |
| 2 | 74213276 | 74213664 | 389 | 25.24755 |
| 3 | 74254659 | 74255045 | 387 | 26.17290 |
| 4 | 74297854 | 74298212 | 359 | 29.61678 |
| 5 | 74298335 | 74298676 | 342 | 32.86220 |
| 6 | 75056785 | 75057221 | 437 | 19.82668 |
| 7 | 75067771 | 75068355 | 585 | 64.71461 |
| 8 | 75579380 | 75579621 | 242 | 13.30696 |
Таблица 1. Информация о найденных пиках.
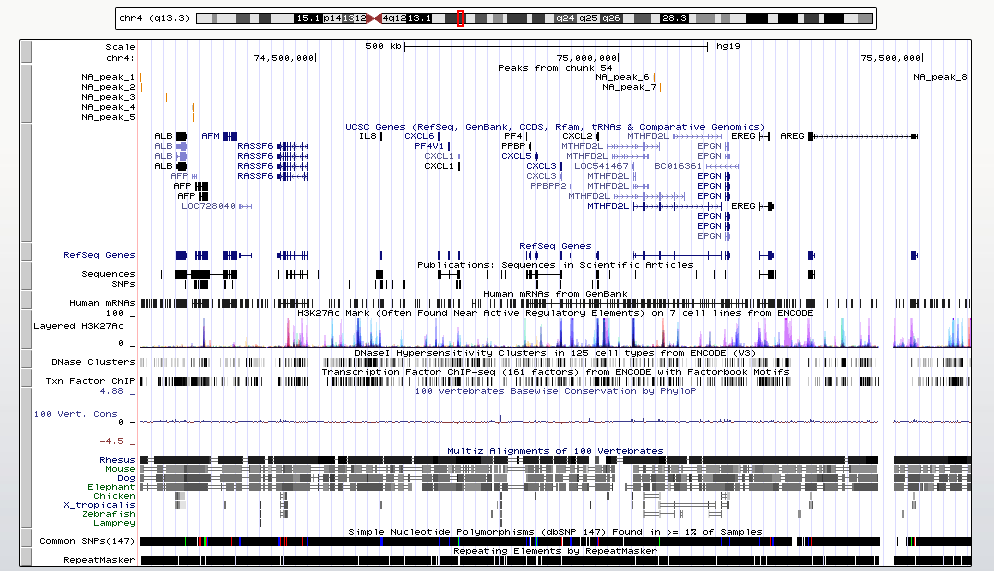
Рис.3 Визуализация пиков в UCSC Genome Browser
Каждому найденному пику соответствует определенное число, позволяющее оценить достоверность находки, — -log10(p-value).
При этом чем больше это число, тем меньше p-value и, следовательно,
тем выше достоверность находки. В принципе, у всех находок
достаточно низкий p-value. Тем не менее я решила рассмотреть пятую и седбмую находки, как наиболее статистически достоверные.
На рис. 4 и 5 представлены изображения двух этих пиков в UCSC Genome Browser.
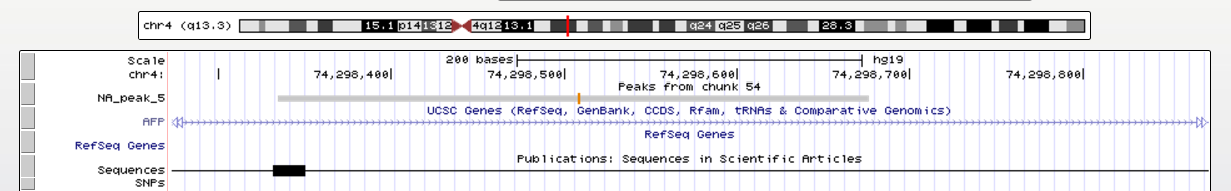
Рис.4 Визуализация пятого пика в UCSC Genome Browser
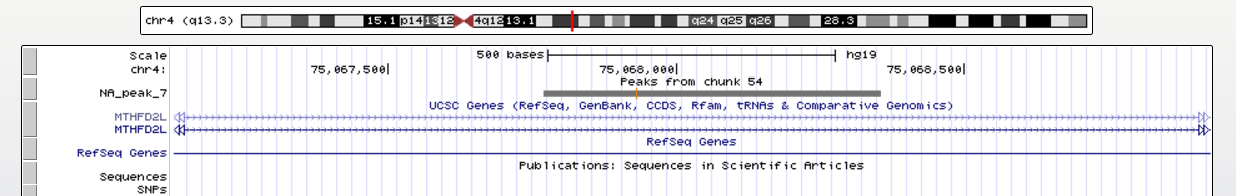
Рис.5 Визуализация седьмого пика в UCSC Genome Browser
Как можно заметить, пятый пик попал на ген AFP, а седьмой - на ген MTHFD2L, а точнее на их интроны.
Ген AFP кодирует белок альфа-фетопротеин, основной белок плазмы, продуцируемый желточным мешком и
печенью во время внутриутробного развития. Белок является эмбриональным аналогом сывороточного альбумина,
а гены альфа-фетопротеина и альбумина находятся в одной и той же транскрипционной ориентации на хромосоме 4.
Альфа-фетопротеин обнаружен в мономерных, а также в димерных и тримерных формах и связывает медь, никель,
жирные кислоты и билирубин.
Ген MTHFD2L кодирует фермент метилтетрагидрофолат дегидрогеназу, которая участвует в одноуглеродном метаболизме (One-carbon metabolism),
катализируя следующие реакции:
5,10-methylenetetrahydrofolate + NAD+ = 5,10-methenyltetrahydrofolate + NADH.
5,10-methenyltetrahydrofolate + H2O = 10-formyltetrahydrofolate.
В качестве кофактора выступает Mg2+
Пики целиком попадают на интрон.
Далее при помощи команды:
/srv/databases/ngs/tools/seqtk/seqtk subseq /srv/databases/ngs/hg19/GRCh37.p13.genome.fa NA_peaks.narrowPeak > my_peaks.fa
был получен файл в формате .fasta, последовательности из которого затем были выровнены в программе Jalview (Muscle with Defaults).
Полученное выравнивание представлено на рис. 6.
 |
Как видно на изображении, в выравнивании очень много гэпов и мало консервативных позиций.
Определить консенсусную последовательность длиной 4-8 нуклеотидов и с
хотя бы 70% консенсуса для каждой позиции не представляется возможным. Последовательности были даны на вход программе МЕМЕ. И действительно, хоть программа и смогла
найти несколько мотивов, но все находки обладали огромным e-value (все были > 1), и достоверными считаться никак не могли.
Вероятно, поскольку экспериментальных данных было недостаточно, не удалось определить последовательности мотивов достаточно точно.
Источники:
© Avdiunina Polina, 2017