
Рис.1 Фрагмент доменных архитектур белков, содержащих домен AMP-binding_C_2
Для работы был выбран домен AMP-binding_C_2 (AC: PF14535) - один из белков, выданных для работе в первом семестре.
Этот домен найден у 1319 видов в составе 2078 последовательностей белков,
и всего для него известно 11 архитектур.
Белки, содержащие этот домен, включают фенилацетат-кофермент А лигазу, которая
катализирует активацию фенилуксусной кислоты
в фенилацетил-КоА [PubMED: 8969218, PubMED: 12189419]. Домен представляет собой центральный бета-лист, фланкированный по бокам альфа-спиралями.
Имеет АМФ-связывающую активность, полный список доменных архитектур можно посмотреть по этой ссылке.
Типичный фрагмент
любой архитектуры из списка представлен следующими доменами:
Рис.1 Фрагмент доменных архитектур белков, содержащих домен AMP-binding_C_2
Функционально оба домена отвечают за связывания белка с АМФ, который может выступать в качестве вторичного мессенджера и участвует
в различных биохимических и сигнальных каскадах.
Для дальнейшей работы были выбраны 2 архитектуры, представленные в таблице 1.
| № | Доменный состав | Число последовательностей | Изображение |
|---|---|---|---|
| 1 | AMP-binding и AMP-binding_C_2 | 1984 | |
| 2 | AMP-binding_C_2 | 81 |  |
Таблица 1. Выбранные для работы архитектуры.
С помощью Jalview было открыто полученное выравнивание доменных участков всех белков, содержащих исследуемый домен (раскраска Clustalx).
Также была добавлена 3D структура домена PF14535.
Для этого последовательность B4E7B5_BURCJ была связана с PDB кодом 2Y27.
Узнать, есть ли в данном случае 3D структура одного домена, а также увидеть соответствие между Uniprot ID и PDB кодами можно было в разделе Structures.
Ссылка на Jalview-проект: pro_1.jvp.
Ссылка на выравнивание в формате fasta: ali_1.fasta
Jalview не смог сохранить изображение выравнивания (оно слишком большое), но можно отметить, что в выравнивании очень много гэпов,
но есть немало консервативных позиций и 2
блока(17:26 и 240-248).
Далее с помощью скрипта swisspfam_to_xls.py были отобраны последовательности
с моим доменом из файла /srv/databases/pfam/swisspfam.gz, содержащего информацию об архитектуре
всех последовательностей:
Команда: python swisspfam-to-xls.py -z -i swisspfam.gz -p PF00569 -o archs.xls
Для полученной таблицы была составлена сводная таблица, в которой столбцами являются разные домены, а строками — идентификаторы белков.
Исследуемый домен выделен красным, домен AMP-binding
(AC: PF00501) - зеленым. Затем для всех идентификаторов были скачаны соответствующие последовательности (Uniprot > Retrieve),
для которых была получена таксономия с помощью скрипта
uniprot_to_taxonomy.py:
Команда: python uniprot-to-taxonomy.py -i uniprot.txt -o tax.xls
Информация по таксономии была добавлена в сводную таблицу, также отдельным скриптом на Python была получена длина домена и добавлена в отдельную колонку.
Итоговая таблица (см. лист "Итоговая таблица").
С помощью несложного скрипта на Python был получен файл ids.txt с исходными идентификаторами.
Затем с помощью скрипта filter-alignment.py были оставлены только выбранные последовательности:
Команда: python filter-alignment.py -i align.fa -m ids.txt -o align_selected.fa -a "_"
Полученный файл с выравниванием доменов выбранных последовательностей был открыт в Jalview, была проведена чистка выравнивания: были удалены пустые колонки, N- и C-участки;
а потом созданы две группы для архитектур и раскрашены по ClustalX (20%). На рис. 2 представлено полученное выравнивание. Оно также доступно в виде проекта.

Рис.2 Полученное итоговое выравнивание доменов.
На изображении выравнивания видно, что для первой архитектуры последовательности домена содержат несколько
больше консервативных между таксонами позиций (и меньше гэпов), чем для второй.
В целом, в выравнивании
последовательностей со первой архитектурой можно выделить целые довольно большие вертикальные блоки,
в то время как
в последовательностях со второй архитектурой наблюдаются бОльшие различия. Хочу также отметить, что для второй архитектуры
у последовательностей значительно варьируется участок
с 71 по 110 аминокислоту (причем без привязки к таксону). В первом большом блоке
выравнивания также можно заметить бОльшую вариативность участков во второй группе, чем в первой.
Вообще говоря, я бы сказала
, что в обоих случаях вертикальные блоки присутствуют.
Расшифровка кодов архитектур и таксонов: 1 - один домен в архитектуре, AMP-binding_C_2;
2 - два домена в архитектуре, AMP-binding_C_2 и AMP-binding; А - Actinobacteria; P - Proteobacteria;
Для полученного итогового выравнивания
было построено дерево с помощью программы MEGA методом Neighbour-joining с использованием bootstrap (100 реплик). Полученные деревья были сохранены в
формате .nwk:
дерево с длинами ветвей, дерево с бутстрэп-поддержкой ветвей.
Изображение дерева можно увидеть на рис. 3, скобочная формула дерева приведена здесь.
.
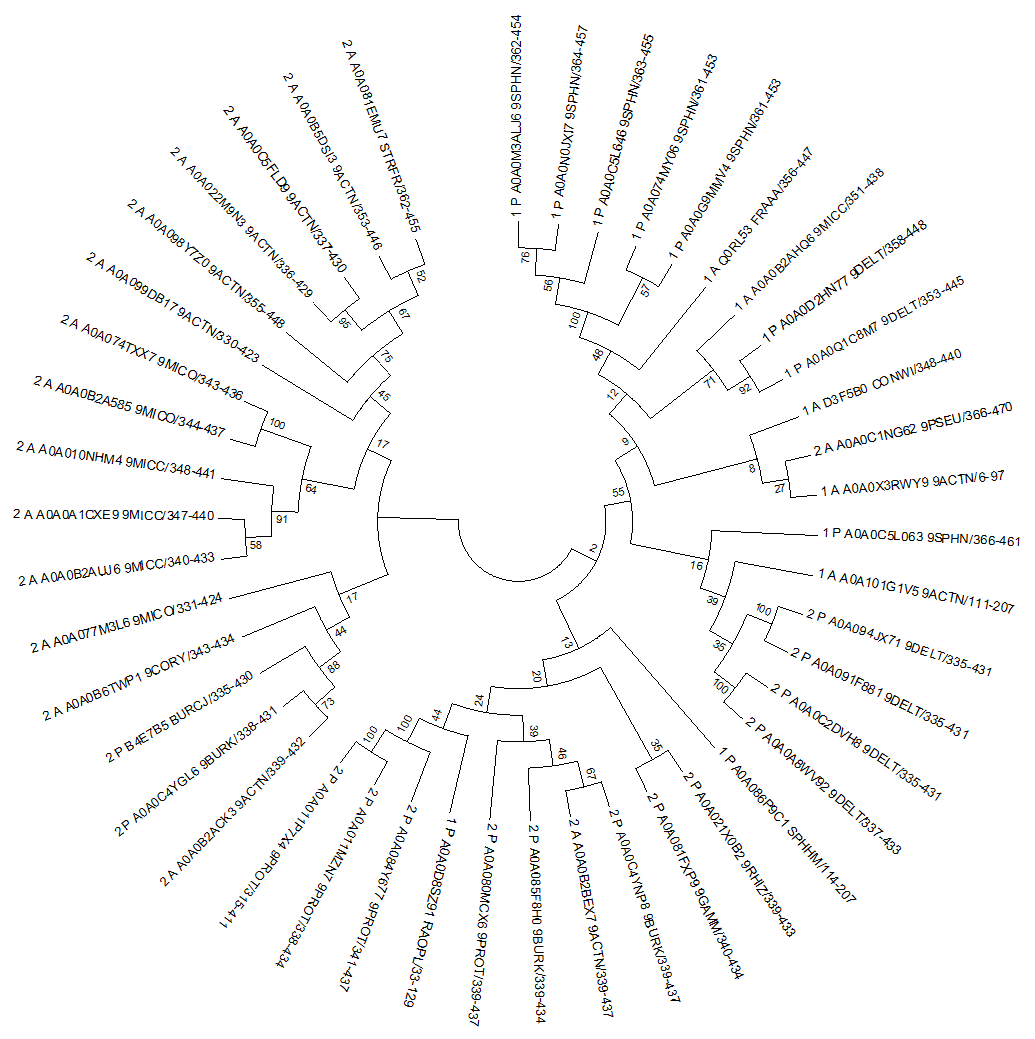
Рис.3 Построенное методом NJ + bootstrap (100 реплик) итоговое дерево.
Как видно на рис.3, дерево разделено на две клады, в одной содержится только вторая архитектура(AMP-binding_C_2 и AMP-binding), в другой обе.
Исходя из выравнивания и из самого дерева
можно сделать вывод, что домены по аминокислотной последовательности очень схожи, что подтверждает их функциональная
синонимичность.
Разделения по таксонам четкого нет: "чужие" последовательности
присутствуют в кладах другого таксона, однако в первой кладе
преобладает таксон Actinobacteria(всего одна последовательность из Proteobacteria).
Таким образом, возможно, у предкового
организма уже существовало две архитектуры для данного домена.
Источники:
© Avdiunina Polina, 2017