ДНК-комплексы с белками
В этом разделе представлена информация о взаимодействиях между ДНК и белком 1RH6.
Для работы в JMol были заданы некоторые множества атомов (см. далее), т.к. они не входят в предопределенные в программе. Это следующие множества:
- множество атомов кислорода 2'-дезоксирибозы (set1),
- множество атомов кислорода в остатке фосфорной кислоты (set2),
- множество атомов азота в азотистых основаниях (set3).
Был создан скрипт-файл script-sets.txt, вызов которого в JMol дает последовательное изображение всей структуры, только ДНК в проволочной модели (рис 1.), той же модели, но с выделенными шариками множеством атомов set1, затем set2 и set3 (рис. 2).
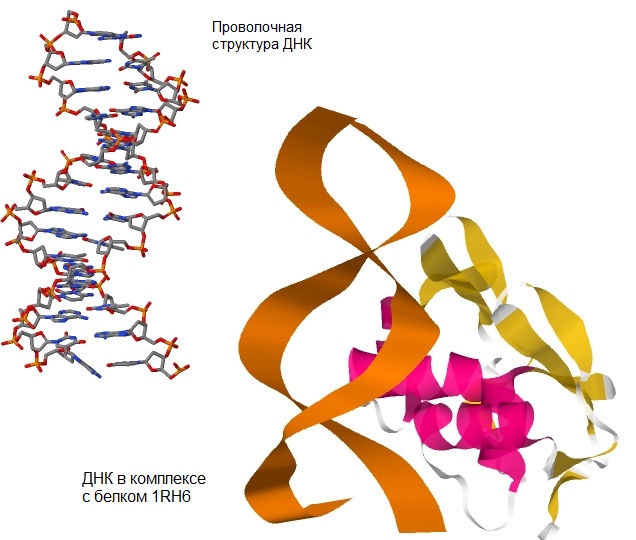
Рис. 1. 1RH6: ДНК в комплексе с белком, проволочная модель ДНК.
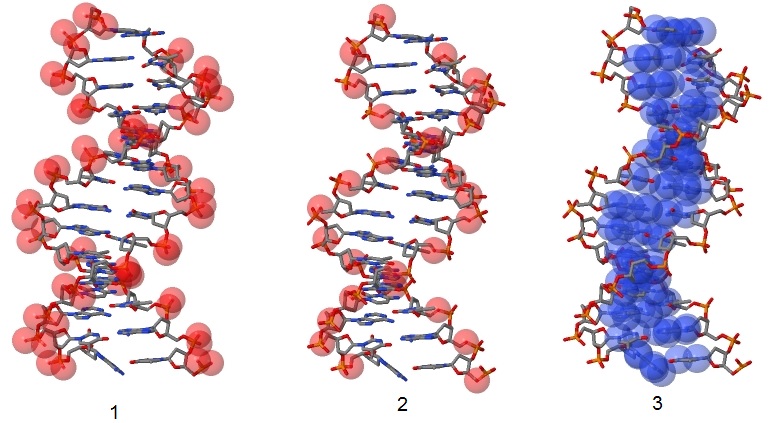
Рис. 2. 1RH6: ДНК с выделенными группами атомов set1-3 (1, 2, 3 соответственно) (пояснения в тексте).
ДНК-белковые контакты
Будем считать полярными атомы кислорода и азота, а неполярными - атомы углерода, фосфора и серы. Назовем полярным контактом ситуацию, в которой расстояние между полярным атомом белка и полярным атомом ДНК меньше 3.5A. Аналогично, неполярным контактом будем считать пару неполярных атомов на расстоянии меньше 4.5A.
Тогда можно подсчитать число контактов разной природы в структуре (1RH6). Данные получены при помощи JMol с использованием скрипта script-links.txt и представлены в таблице 1.
Таблица 1. Число контактов между различными группами атомов ДНК с белком.
| Контакты атомов белка с | Полярные | Неполярные | Всего |
| остатками 2'-дезоксирибозы | 6 | 26 | 32 |
| остатками фосфорной кислоты | 0 | 17 | 17 |
| остатками азотистых оснований со стороны большой бороздки | 1 | 2 | 3 |
| остатками азотистых оснований со стороны малой бороздки | 1 | 4 | 5 |
Популярная схема ДНК-белковых контактов
Популярная схема ДНК-белковых контактов может быть получена с помощью программы nucplot. Выдачу можно увидеть на рисунке 3.
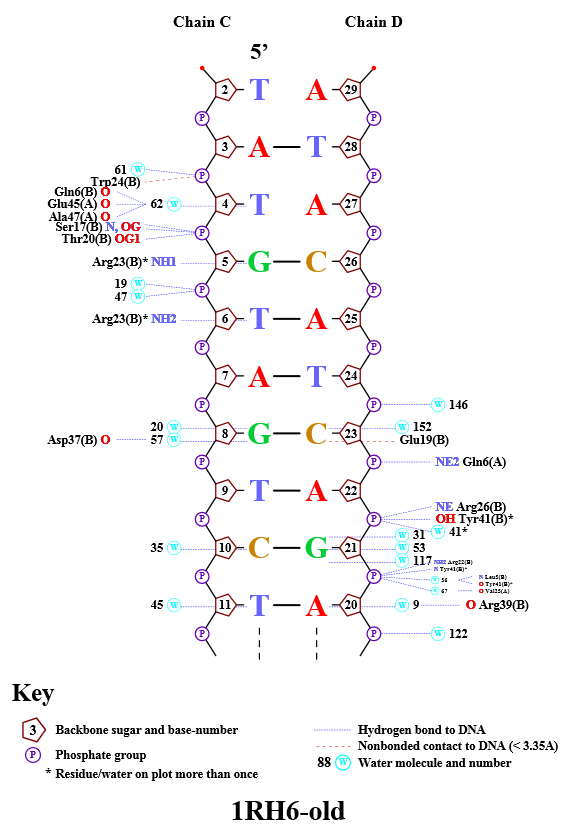
Рис. 3. Схема ДНК-белковых контактов, полученная с помощью программы nucplot.
На схеме значительно меньшее количество ДНК-белковых контактов, чем было найдено с помощью JMol. Вероятно, это связано с тем, что выбранные расстояниями для предполгаемого контакта не всегда соответствует наличию этого контакта.
На схеме видно, что только две аминокислоты - Arg23(B) и Tyr41(B) - имеют по 2 контакта с ДНК (это наибольшее число в данном случае), причем эти контакты представляют собой водородные связи. Выделенные аминокислотные остатки и взаимодействующие с ними (исходя из информации со схемы) нуклеотиды представлены на рисунках 4-5. Использовался скрипт JMol script-links.txt.
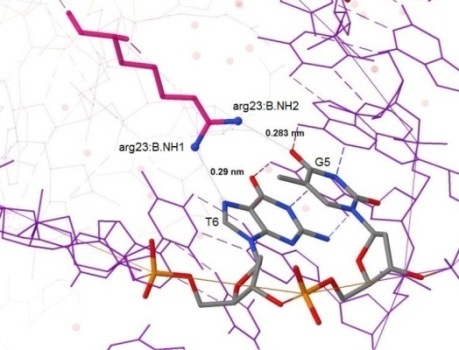
Рис. 4. Остаток arg23(B) и взаимодействующие с ним нуклеотиды T6 и G5. Измерено расстояние предполагаемых (из данных схемы) водородных связей.
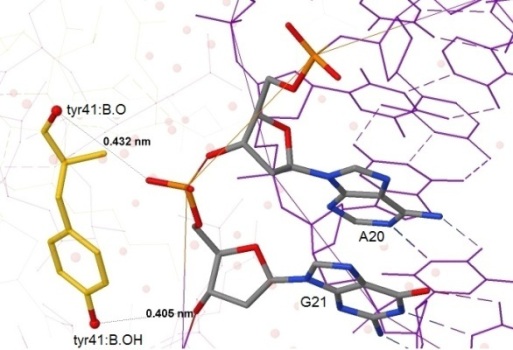
Рис. 5. Остаток tyr41 и взаимодействующие с ним нуклеотиды A20 и G21. Измерены расстояния от контактирующих атомов тирозина (О и ОН) до ближайших: это атомы, принадлежащие G21.
Вывод. Как видно на рисунке 5, расстояния от контактирующих атомов тирозина до ближайших к нему атомов ДНК больше 0.4 нм, значит, водородных связей (о которых говорится на схеме) быть не должно. По крайней мере, из этой информации можно сделать вывод, что важнее для распознавания последовательности ДНК белком более важным является остаток аргинина arg23(B), который образует водородные связи с азотистыми основаниями.
Предсказание вторичной структуры тРНК из 1RTR
I. Предсказание структуры тРНК путем поиска инвертированных повторов.
Программа einverted из пакета EMBOSS позволяет найти инвертированные участки в нуклеотидных последовательностях. Были найдены возможные комплементарные участки в последовательности исследуемой тРНК. Результаты внесены в таблицу 2.
Параметр "Minimum score threshold" для нахождения каких-либо канонических пар пришлось снизить до 15 (от 50 по умолчанию). Найдены были только пары акцепторного стебля тРНК. Результат не изменялся при еще большем уменьшении значения указанного параметра. Выдача программы представлена в файле sequence.inv.
Для запуска программы einverted была использовалась команда:
II. Предсказание структуры тРНК по алгоритму Зукера.
Программа mfold из пакета EMBOSS реализует алгоритм Зукера. Наиболее подходящими параметрами для получения предсказания, наиболее близкого к реальной структуре, оказались параметры по умполчанию. Полученная структура представлена на рисунке 5. Ее можно назвать неудачной, т.к. она предполагает объединение имеющихся T- и D-стеблей тРНК. Но внутри длинного стебля есть несколько неканонических пар. Вероятно, программа считает их наличие недостаточно значимым и рисует стебель.
В то же время, программой найдены все канонические пары (19). Результаты внесены в таблицу 2. Изменение параметров по умолчанию (например, параметра P) приводило к ухудшению структуры, так что можно считать, что наилучшее предсказание было получено с первого раза.
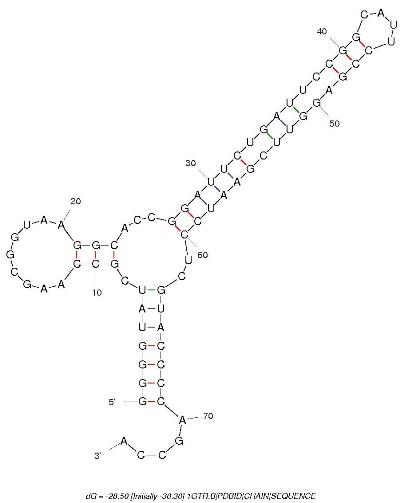
Рис. 5. Структура тРНК из 1GTR.pdb, полученна с помощью алгоритма Зукера с параметрами по умолчанию.
Таблица 2. Количество пар в стеблях тРНК (1GTR) и количество предсказанных пар разными способами.
| Участок структуры | Позиции в структуре (по результатам find_pair) | Количество предсказанных с помощью einverted | Результаты предсказания по алгоритму Зукера |
| Акцепторный стебель | 5'-2-7-3', 5'-71-66-3' (Всего 6 пар) | 6 | 6 (в стебле 1-7, 63-69) |
| D-стебель | 5'-39-43-3', 5'-27-31-3' (Всего 5 пар) | 0 | 10 (в стебле 27-41, 46-60) |
| T-стебель | 5'-49-53-3', 5'-65-61-3' (Всего 5 пар) | 0 | |
| Антикодоновый стебель | 5'-10-12-3', 5'-23-25-3' (Всего 3 пары) | 0 | 3 (в стебле 9-11, 21-23) |
| Общее число канонических пар нуклеотидов | 19 | 6 | 19 |