ФББ 2013-2014


ФББ 2013-2014
Картирование на референсный геном
Возьмём риды из предыдущего практикума, очищенные программой Trimmomatic, и картируем их на референсный геном, в качестве которого выступят хлоропласт и митохондрия Arabidopsis thaliana. Для выравнивания ридов на геном использовалась программа BWA (Burrows–Wheeler alignment), которая работает по алгоритму преобразования Барроуза — Уилера. Этот алгоритм используется для сжатия данных. Во время работы не меняется ни один символ строки, но повторы в подстроках упорядочиваются таким образом, что данные становится легче сжимать. Также по файлу, полученному с помощью этого алгоримта, можно восстановить исходный.
Для картирования произведём сначала индексацию последовательности с помощью программы bwa index sequence.fasta. Само картирование выполняется командой bwa mem sequence.fasta trim.fastq > map.sam. На выходе получается файл в формате .sam (sequence/alignment map), работа с такими файлами осуществляется с помощью пакета samtools.
Теперь нам нужно перевести файл .sam в формат .bam. Bam - бинарный файл, он может весить в разы меньше, чем .sam, поэтому с ним удобнее работать. Перевод осуществляется командой: samtools view -b -S -h map.sam > map.bam . Значение опций: -b - перевести в бинарный файл, -S - на вход подаётся файл .sam, -h - сохранять заголовки.Далее проведём сортировку и индексацию полученного .bam файла следующими командами: samtools sort map.bam map; samtools index map.bam. После этого с файлом можно легко работать не только в samtools, но и в некоторых программах для визуализации картирования (например, в BamView). Выравнивания были визуализированы, чтобы сравнить "на глаз" покрытие и заранее оценить, на что картировалось лучше - на митохондрию или хлоропласт. На рисунке 1 показаны результаты картирования для хлоропласта, график внизу показывает покрытие.

Рис.1. Визуализация картирования на хлоропласт
Видно, что риды откартировались на хлоропласты хорошо, с высоким покрытием. Теперь посмотрим на рисунок 2, где показаны результаты для митохондрии. Уже по картинке можно понять, что хлороплас выиграл.
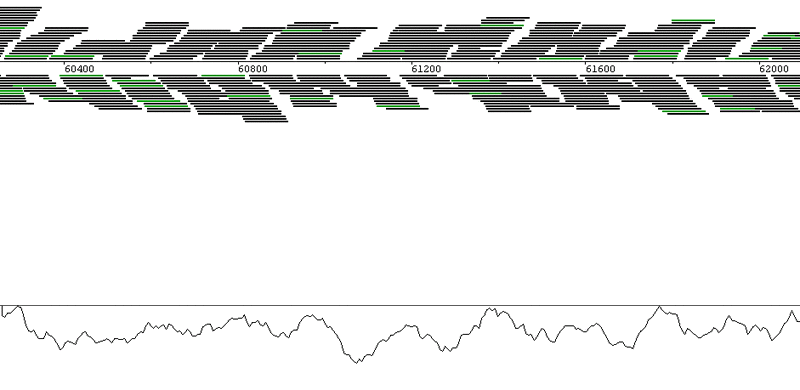
Рис.2. Визуализация картирования на митохондрию
Также можно оценить картирование с помощью программ из samtools. Статистика смотрится с помощью команды: samtools idxstats map.bam. Вот она:
ENA|AP000423|AP000423.1 154478 670023 0 ENA|Y08501|Y08501.2 366924 73370 0 * 0 0 3115103Это значит, что геном хлоропласта был длиной 1544878 нуклеотидов, откартировалось на него 670023 рида, а на геном митохондрии, длиной 366924 нуклеотида, откартировалось 73370 ридов. Очевидно, что хлоропластов в исходном образце было больше, что логично для растения.
Теперь проверим покрытие каждого нуклеотида командой: samtools depth map.bam > coverage.txt. Видно, что разброс покрытий очень велик, однако на хлоропласт (идентификатор AP000423.1) покрытие в среднем выше, чем на митохондрию (идентификатор Y08501.2). Оценить эту разницу вы можете, посмотрев файл с покрытиями.