ФББ 2013-2014


ФББ 2013-2014
A-, B- и Z- формы ДНК
С помощью пакета 3DNA были построены A, B и Z формы ДНК. A и B состоят из GATC повторов, Z - только из GC пар. Использованная команда: fiber -(a,b,z) filename.pdb
На рисунках 1, 2 и 3 показаны формы ДНК, построенные программой. Скачать полученные pdb файлы можно по ссылкам :
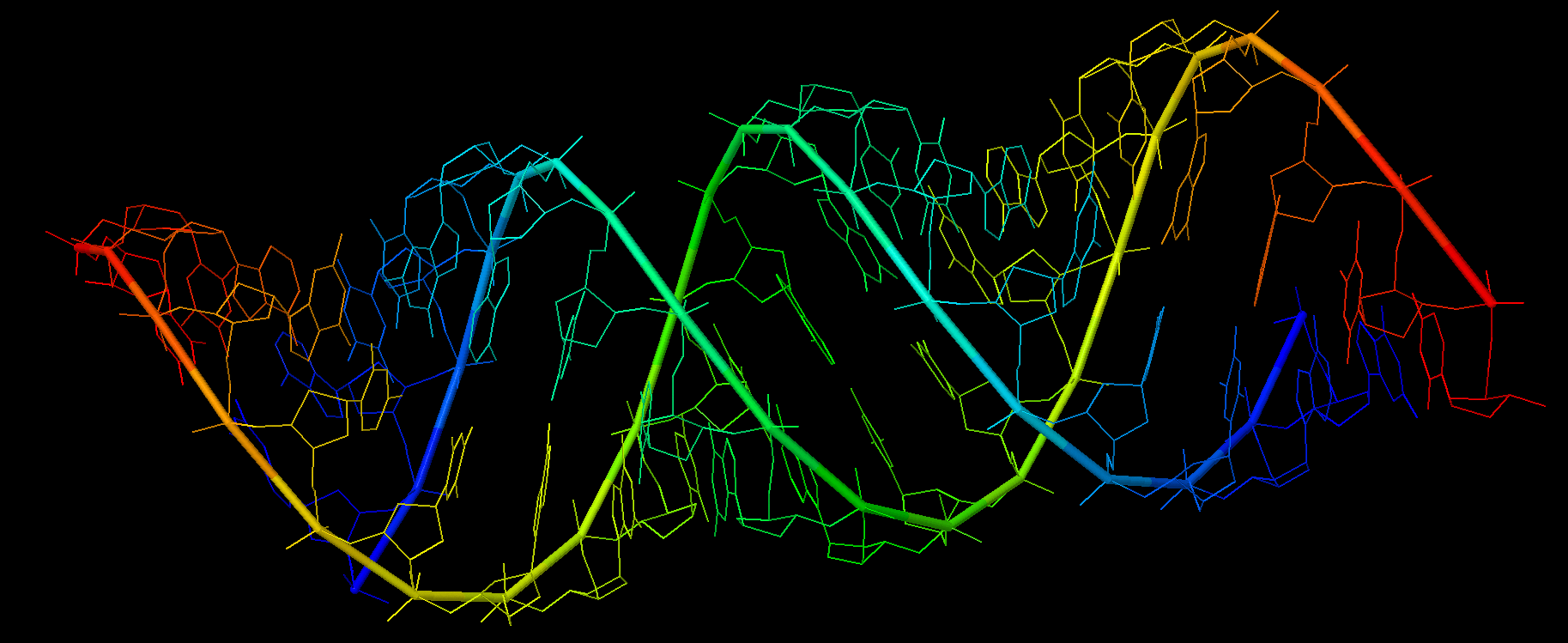

Рис. 1. А форма ДНК. Изображения в профиль и с торца. Покраска color group в Jmol.
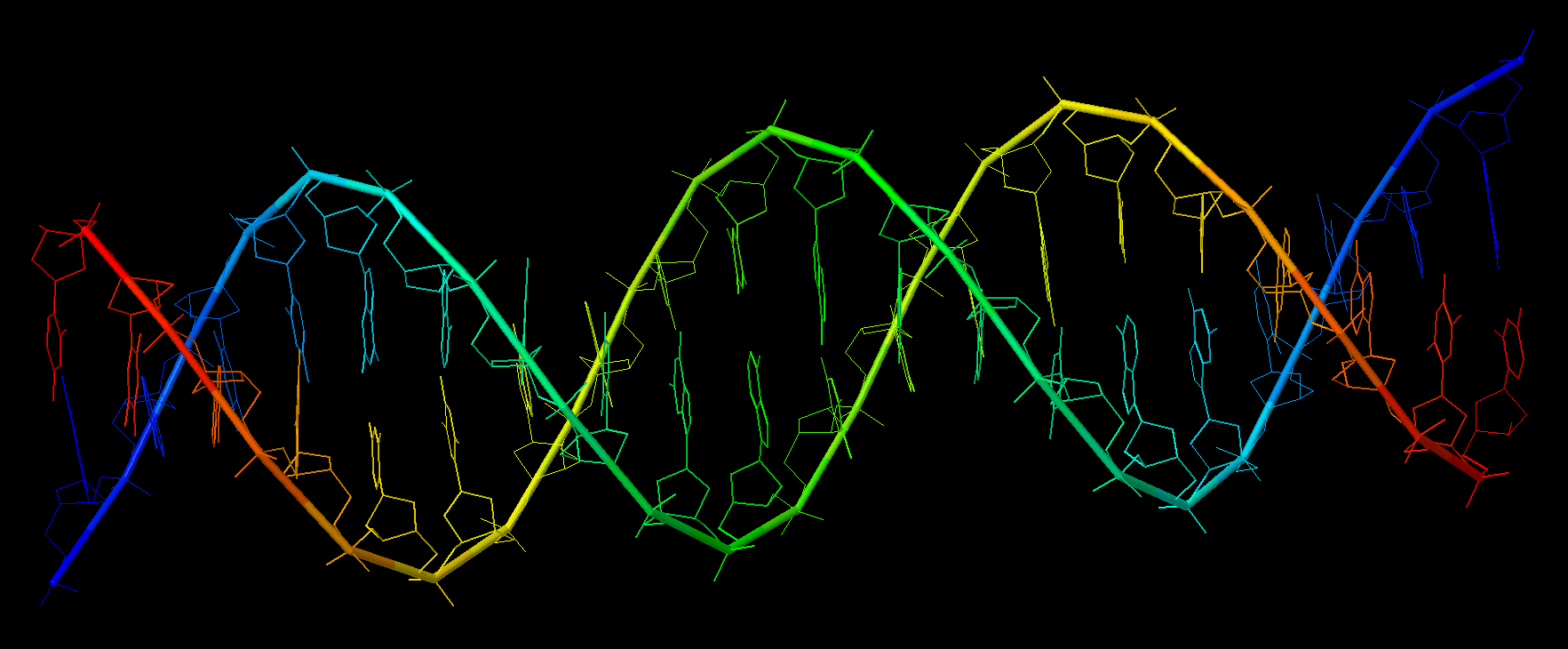
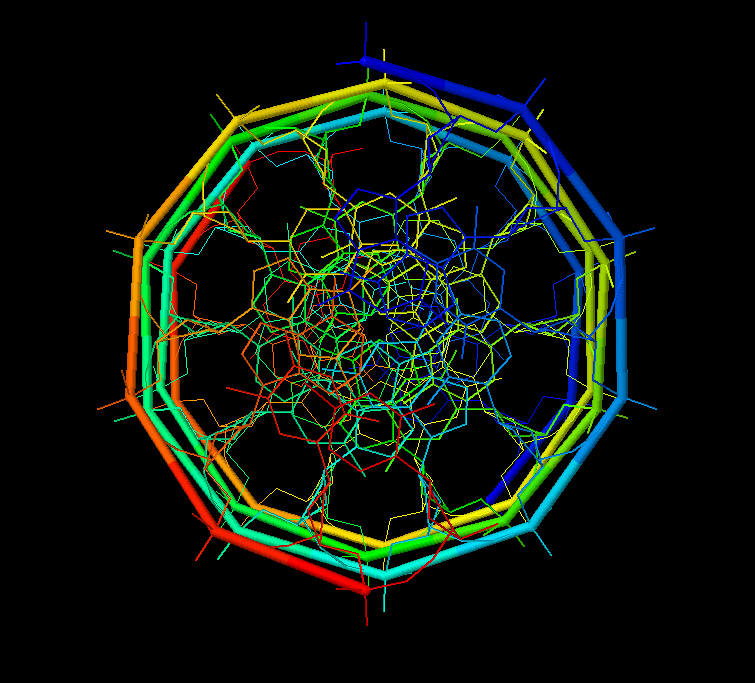
Рис. 2. B форма ДНК. Изображения в профиль и с торца. Покраска color group в Jmol.
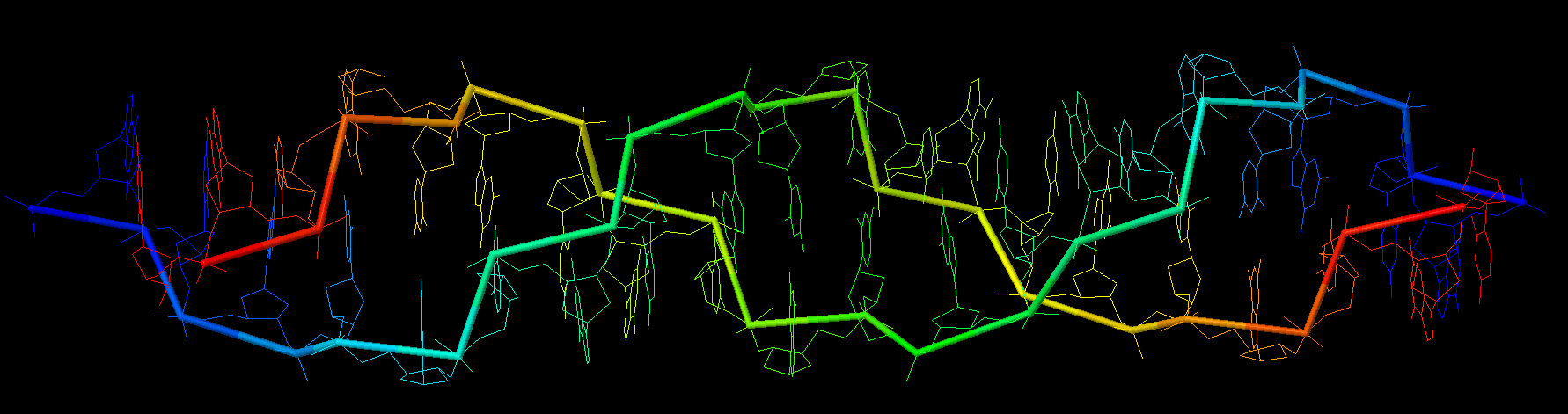

Рис. 3. Z форма ДНК. Изображения в профиль и с торца. Покраска color group в Jmol.
Задание 1: используя pdb файл А формы, выделим сахарофосфатный остов (рис 4), все нуклеотиды (рис. 5), все аденины (рис. 6), атом N7 в первом гуанине (рис.7).

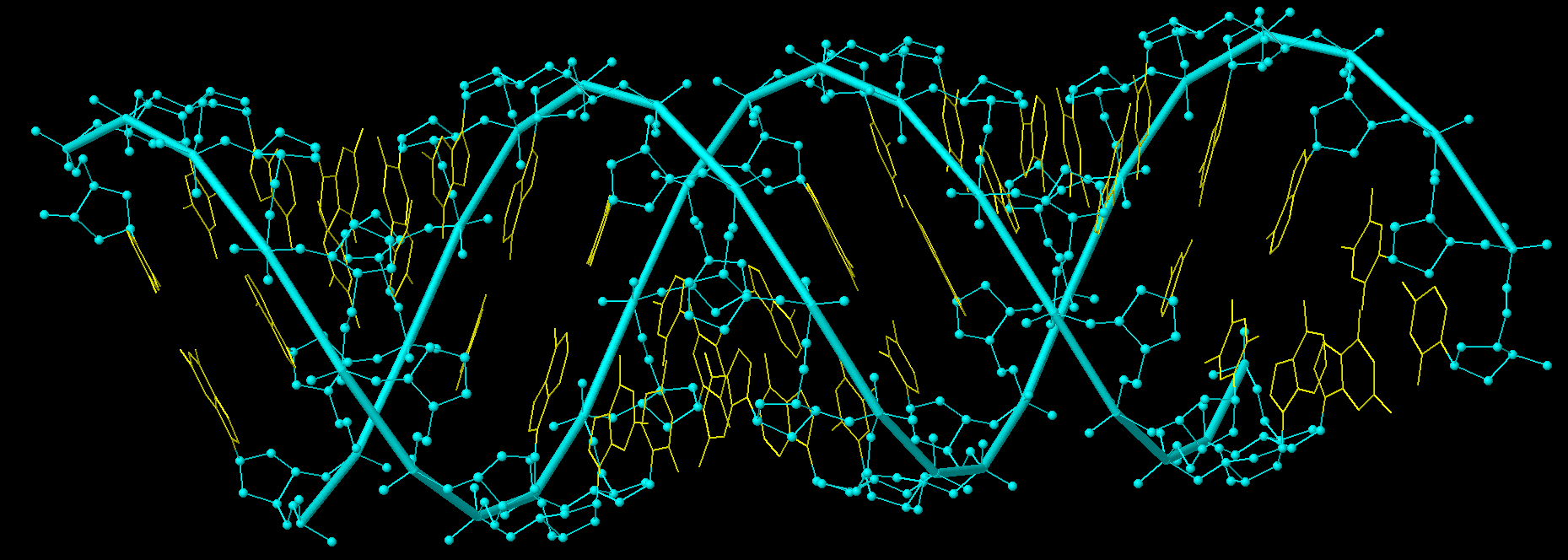
Рис. 4. Сахарофосфатный остов выделен голубым цветом, всё остальное - жёлтым. Команда в Jmol - select backbone

Рис. 5. Выделены цветом все нуклеотиды (то есть вся ДНК, т.к. ДНК состоит из нуклеотидов).

Рис. 6. Жёлтым цветом выделены аденины. Команда в Jmol - select A

Рис. 7. Выделенный жёлтым атом - седьмой азот у первого гуанина. Модель cpk.
Задание 2: было дано 2 PDB файла, в одном из них был комплекс РНК+белок, в другом - ДНК+белок. Скачать данные pdb файлы можно по ссылкам 1PP8 (ДНК+белок), 1GTS (РНК+белок). На рисунках 8 и 9 представлены изображения данных комплексов.
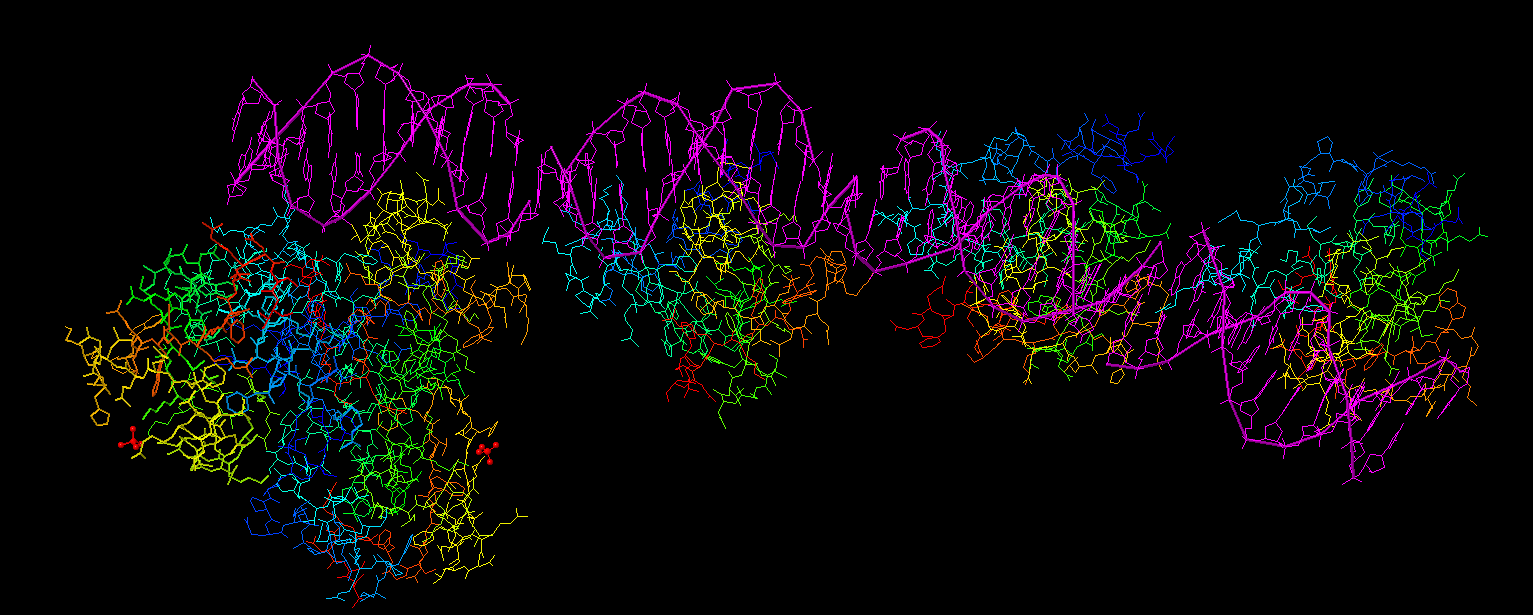
Рис. 8. 1GTS - комплекс ДНК+белок.Малиновым выделена нуклеиновая кислота, субъединицы белка - радужная раскраска.
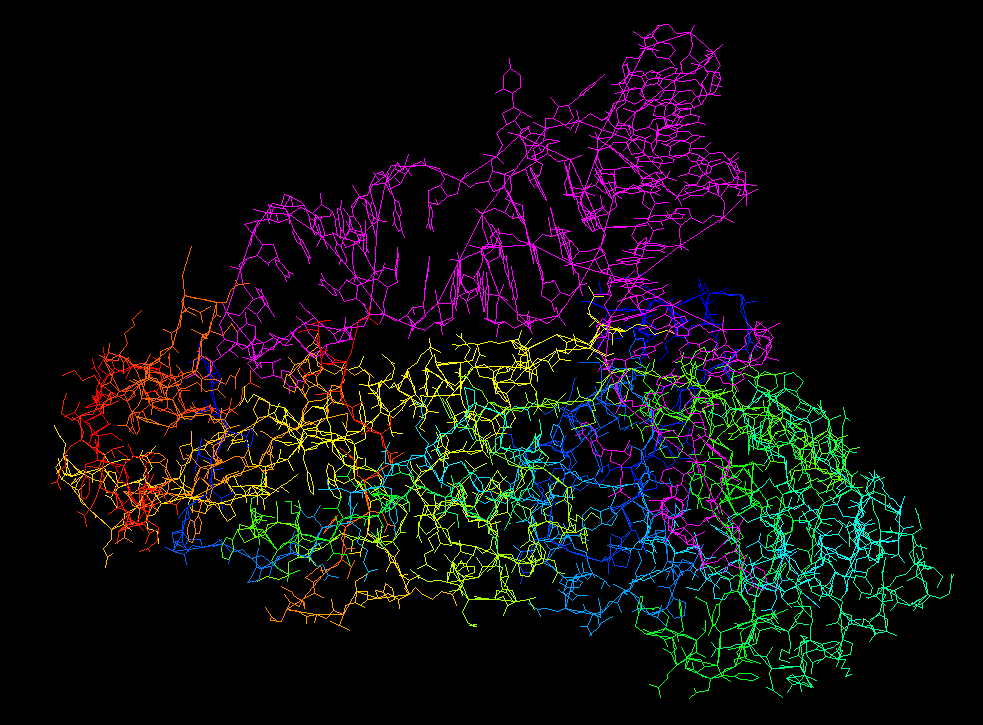
Рис. 9. 1PP8 - комплекс РНК+белок.Малиновым выделена нуклеиновая кислота, субъединицы белка - радужная раскраска.
Далее было необходимо проверить структуры нуклеиновых кислот на наличие разрывов. В данной РНК разрывов найдено не было. В ДНК же ситуация обратная, разрывов много. Это могло произойти по причине недостаточного разрешения рентгеноструктурного анализа. Это предположение было сделано на основе того, что разрывы расположены симметрично и с определённой периодичностью. Но это также может указывать на то, что механизм работы белка включает в себя разрезание ДНК. Но это вряд ли.
Результаты представлены на рисунках 10 и 11.

Рис. 10. Структура ДНК из данного файла. Разрывы обведены в белые овалы.
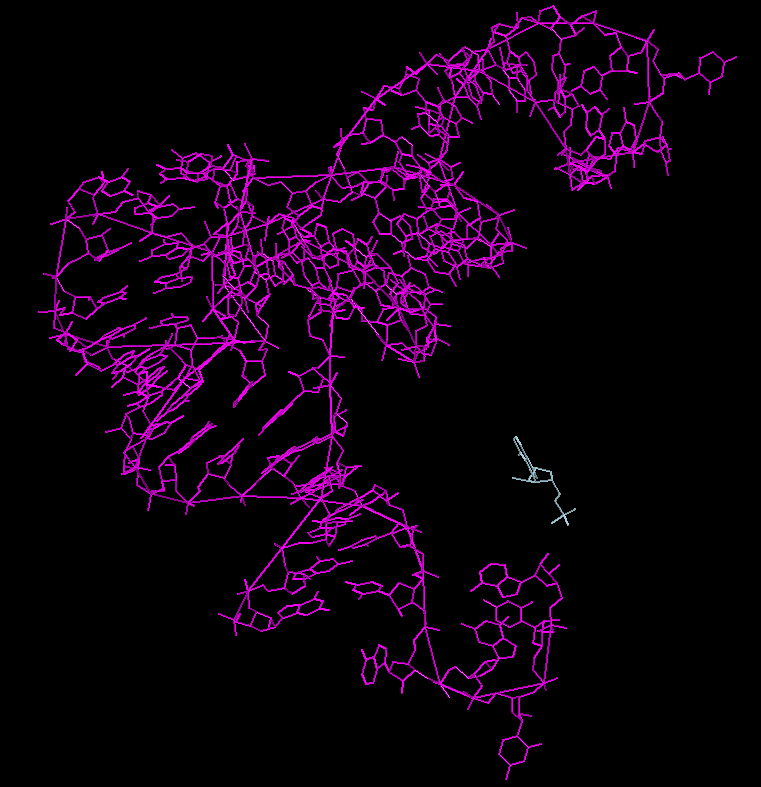
Рис. 11. РНК, видимых разрывов нет. Голубым цветом показан АМФ.
В задании 3 было необходимо найти большие и малые бороздки в разных формах ДНК. Начнём с наиболее распространённой формы - B-формы. На рисунке 12 белой и розовой линией показаны большая и малая бороздки соответственно. В В-форме их размер соответствует названиям, однако в других формах большая бороздка не всегда больше малой. Главное отличие двух бороздок друг от друга в глубине: большая бороздка всегда глубже малой. Определение бороздок важно, так как многие ферменты взаимодействуют именно с большой бороздкой ДНК, потому что нуклеотиды в ней лежат более открыто.
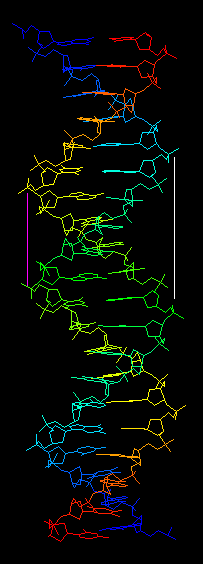
Рис. 12. B-спираль ДНК с обозначенными большой (белая линия) и малой (розовая линия) бороздками.
Атомы оснований, которые составляют ДНК, можно экспонировать в большую или малую бороздки. Проделаем это для тимина. Рисунок 13, полученный с помощью ChemSketch, отражает положение атомов относительно бороздок (красным цветом показаны атомы, обращённые в большую бороздку; синим - в малую).

В сторону малой бороздки обращены N1, C2, O2. В сторону большой бороздки обращены С4, С5, С6, С5М, О4 Остальные атомы - N3
Теперь перейдём к рассмотрению и других форм ДНК. В таблице 1 представлены характеристики всех трёх форм, все данные были получены с помощью Jmol, процесс измерения показан на рисунке 14.
Таблица 1. Сравнение характеристик 3х наиболее распространённых форм ДНК
| A-форма | B-форма | *Z-форма | |
| Тип спирали (правая или левая) | Правая | Правая | Левая |
| Шаг спирали (нм) | 2.803 (от P1) | 3.091 (от P1) | 4.35 (от P1) |
| Число оснований на виток | 12 | 10 | 13 |
| Ширина большой бороздки (нм) | 0.798 | 1.791 | 0.72 |
| Ширина малой бороздки (нм) | 1.697 | 1.169 | 1.517 |
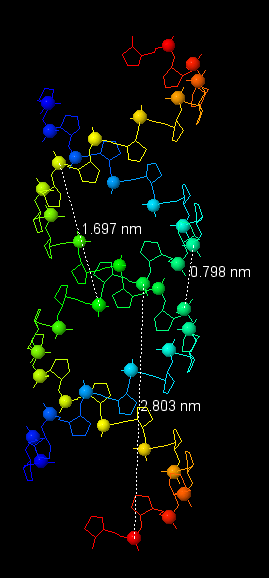
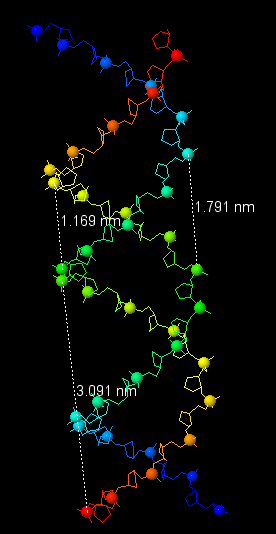
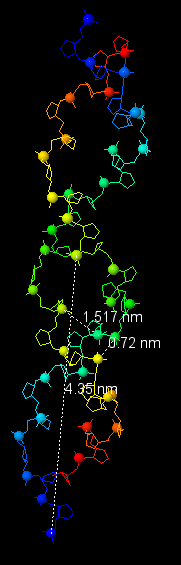
Рис. 14. Измерения шага спирали и длины бороздок для всех трёх форм ДНК.
Теперь измерим торсионные углы для Т11. Это возможно сделать с помощью средств Jmol. В нуклеиновых кислотах обычно измеряют 7 таких углов, для их измерения необходимо 4 атома. Таблица, в которой показано, между какими атомами мы будем измерять торсионные углы, приведена ниже (составлено не мной):
alpha: O3'(i-1)-P-O5'-C5'
beta: P-O5'-C5'-C4'
gamma: O5'-C5'-C4'-C3'
delta: C5'-C4'-C3'-O3'
epsilon: C4'-C3'-O3'-P(i+1)
zeta: C3'-O3'-P(i+1)-O5'(i+1)
chi for pyrimidines(Y): O4'-C1'-N1-C2
chi for purines(R): O4'-C1'-N9-C4>
Процесс измерения для А-формы показан на рисунке 15, а в таблице 2 приведено сравнение полученных значений торсионных углов для А и В формы со значениями, которые были в презентации.
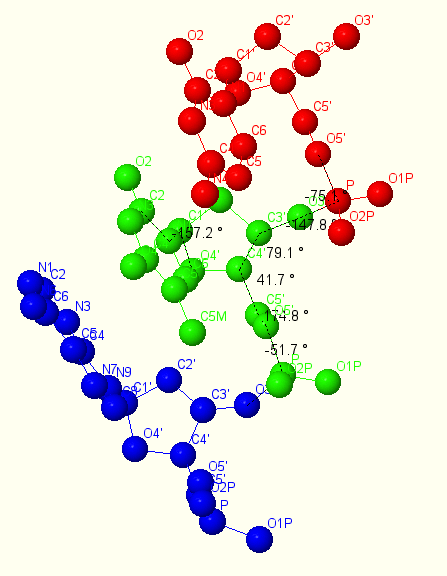
Рис. 15. Измерение торсионных углов для А формы ДНК.
Таблица 2. Полученные торсионные углы для А и В формы ДНК и их сравнение со значениями из презентаций.
| α | β | γ | δ | ε | ζ | χ | |
| A-ДНК (измеренное) | -51.7 | 174.8 | 41.7 | 79.1 | -147.8 | -75.1 | -157.2 |
| А-ДНК (презентация) | 62 | 173 | 52 | 88 | 178 | -50 | -160 |
| B-ДНК (измеренное) | -29,9 | 136,4 | 31.1 | 143.4 | -140.8 | -160.5 | -98 |
| B-ДНК (презентация) | 61 | 171 | 54 | 123 | 155 | -90 | -117 |
Как видно, значения торсионных углов, измеренные вручную и из презентации различаются довольно сильно. Возможно, это произошло потому, что измерения вручную проводились только для тимина, а в презентации были представлены некие обощённые данные (средние значения) для всех нуклеотидов и для природных молекул ДНК.
Теперь сделаем расчёт торсионных углов при помощи программы 3DNA. Сначала переведём наши pdb файлы в старый pdb формат, так как программа работает только с ним. Сделать это можно при помощи программы remediator --old 'XXXX.pdb' > 'XXXX_old.pdb'.
С помощью программ find_pair и analyze получим данные для торсионных углов: составляем из них конвеер find_pair -t gatc_x.pdb stdout | analyze. На выходе получаем множество файлов, нас интересует файл с расширением .out. Открыв его, мы увидим много различных таблиц по различным характеристикам ДНК, нас интересует таблица с торсионными углами. Проделываем это для всех трёх форм ДНК. Значения измеренных торсионных углов для А и В форм совпали с теми, что выдала программа. Для Z формы было обнаружено, что торсионные углы при цитозине и гуанине сильно отличаются друг от друга (это продемонстрировано на рисунке 16).
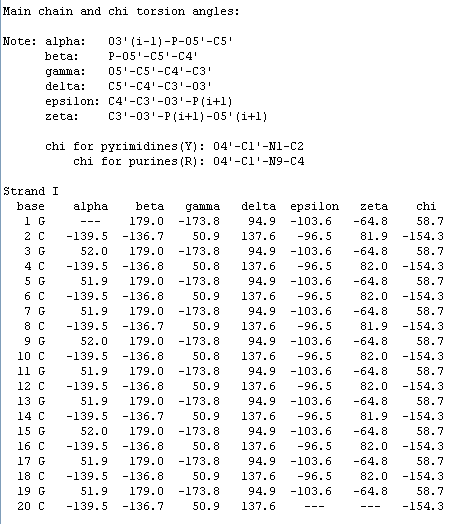
Рис. 16. Измерение торсионных углов для Z формы ДНК с помощью программы 3DNA.
Проанализируем торсионные углы в тРНК из 1GTS.pdb. С помощью Excel были посчитаны средние значения для торсионных углов. Результаты представлены в таблице 3. В сравнении с ДНК, углы тРНК больше похожи на углы А-формы, чем на углы В-формы. Углы для некоторых нуклеотидов сильно отклоняются от средних значений. Это происходит либо в том месте, где вторичная структура формирует изгиб, либо у тех нуклеотидов, которые не могут образовать классическую уотсон-криковскую пару с соседним нуклеотидом (например, один урацил с нестандартными углами лежал напротив другого урацила). Также экстремальные значения торсионных углов могут быть связаны с ошибками при расшифровке структуры.
Таблица 3. Средние значения торсионных углов в тРНК из 1GTS.
| α | β | γ | δ | ε | ζ | χ | |
| Т-РНК | -28,24 | 57,02 | 59,18 | 87,07 | -125,9 | -79,97 | -133,09 |
| Отклоняющийся нуклеотид (G4) | 171,8 | -172,1 | 172,4 | 81,2 | -131,2 | -73,7 | -176,5 |
Теперь проанализируем схожим образом торсионные углы в ДНК из 1PP8.pdb. По измеренным средним значениям сложно судить о том, в какой форме находится ДНК. Результаты в таблице 4.
| α | β | γ | δ | ε | ζ | χ | |
| ДНК из 1PP8.pdb | -54,9 | 30,45 | 34,70 | 135,53 | -141,44 | -100,24 | -123,78 |
| Отклоняющийся нуклеотид (A46) | -179,7 | 154,9 | 158,3 | 138,5 | -150,5 | -97,1 | -145,4 |
Проанализируем вторичную структуру РНК из файла 1GTS.pdb. Известно, что у РНК намного больше разнообразных элементов во вторичной структуре, и она устроена сложнее, чем у ДНК. Выделяют стебли, узлы, псевдоузлы, выпетливания, шпильки и т.д. РНК бывают различных видов и выполняют в клетке большой спектр функций. В нашем случае работа будет проводиться на тРНК. В структуре тРНК выделяют 3 петли (Т-петля, Д-петля и антикодоновая петля), акцепторный стебель. Если нарисовать тРНК схематично, то получится нечто, похожее на клеверный лист.
Для определения стеблей вручную вернёмся к файлу 1GTS.out. Искать будем 4 стебля. Результат представлен на рисунке 17.

Рис. 16. Выделение 4х стеблей в данной тРНК. Красный - акцепторный стебель, голубой - антикодоновый стебель, зелёный - Т-стебель, розовый - Д-стебель.
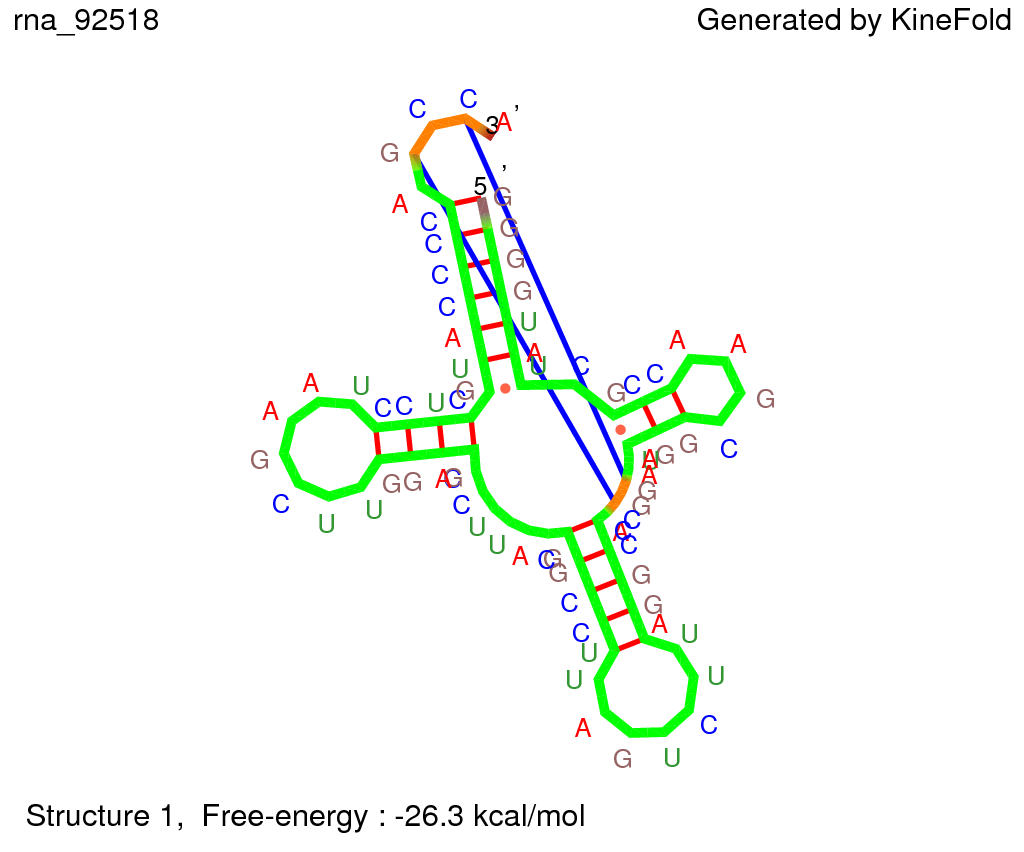
Рис. 17. Структура данной последовательности тРНК, построенная с помощью Kinefold.
Однако в РНК возможны не только канонические взаимодействия нуклеотидов, но и неканонические. По рисункам 16 и 17 можно понять, что это U55:G18, U38:U32, C44:a26, A13:A45. Итого 4 неканонические пары.
Теперь перейдём к рассмотрению стэкинг-взаимодействий. Стэкинг - это явление взаимодействия ароматических колец между собой, которое повышает общую устойчивость соединения. В нуклеиновых кислотах стэкинг параллельный: ароматические кольца нуклеотидов находятся строго друг под другом. На рисунке 18 представлены все стэкинг взаимодействия между основаниями в РНК из 1GTS.pdb. В розовый прямоугольник взяты нуклеотиды, дающие наибольшую площадь перекрывания. В голубых прямоугольниках - пары с наименьшей площадью перекрывания. Как видно, у двух пар вообще нет перекрывания, значит между ними никакого стэкинга не возникает.

Рис. 18. Информация о площади перекрывания, полученная из файла 1GTS.out.
С помощью команд " ex_str -N stacking.pdb stepN.pdb" и "stack2img -cdolt stepN.pdb stepN.ps" были получены изображения пар нуклеотидов с наибольшим и наименьшим перекрыванием. Эти же пары обведены на рисунке 18 в рамочки. На рисунке 19 изображены 3 пары с наименьшей площадбю перекрывания, на рисунке 20 - пары с наибольшим перекрыванием.
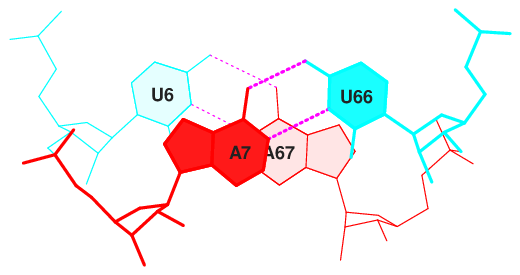

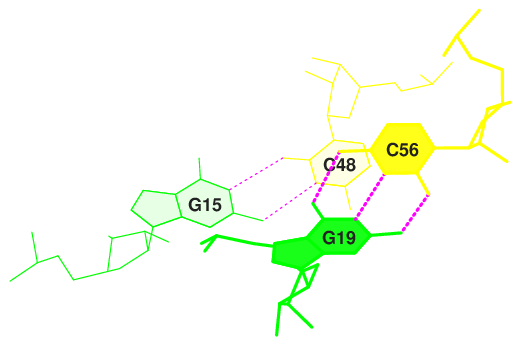
Рис. 19. Пары с наименьшей площадью перекрывания

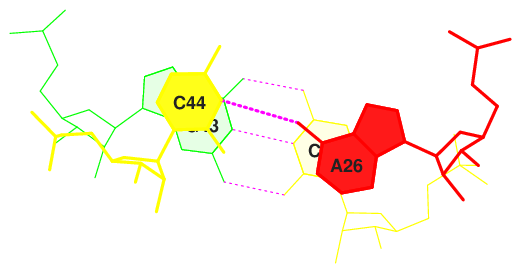
Рис. 20. Пары с наибольшей площадью перекрывания