ФББ 2013-2014


ФББ 2013-2014
Предсказание вторичной структуры тРНК
Для предстакания вторичной структуры РНК есть несколько способов. Один из них - поиск инвертированных участков последовательности с помощью команды einverted (пакет EMBOSS). Попробуем получить наиболее близкое к реальности "выравнивание". Подаём на вход команде файл с последовательностью в формате .fasta. Если оставить стандартные параметры, то в выходном файле будет пустота, поэтому для начала попробуем поставить минимальный штраф за гэпы. Результат:
SEQUENCE: Score 72: 24/24 (100%) matches, 20 gaps
1 ggggt------a-tcgccaag---cggtaag-gcaccgg-attc 32
||||| | ||| || ||| || | |||| | ||
69 ccccatgctcctaagc--tt-ggagcc-tt-ac--ggcctt-ag 34
Очевидно, что эта стратегия слишком проста для получения правильного результата. Теперь попробуем поставить ненулевой штраф за
гэпы, минимизировать minimum score treshold, максимизировать match_score. На выходе мы получим:
SEQUENCE: Score 216: 24/28 ( 85%) matches, 12 gaps
1 ggggtatc---g---ccaa--gcggtaaggcaccggattc 32
|||||| | | | || ||| || | ||||| ||
69 ccccat-gctcctaagcttggagcc-ttac--ggccttag 34
Это уже больше похоже на результаты, полученные в практикуме 2. Однако, как мы видим, нормально просчитан только акцепторный стебель, другие
стебли имеют вставленные гэпы или не совпадают вовсе. Это значит, что пользоваться этой программой для предсказания структуры тРНК не совсем
надежно. Также программа не учитывает неканонических взаимодействий, что также может стать проблемой, так как неканонические взаимодействия
встречаются в тРНК довольно часто.
Теперь применим алгоритм Зукера с той же целью. Я воспользовалась онлайн-сервисом по сворачиванию РНК, который находится здесь. Сначала я получила структуру с наименьшей энергией гиббса (то есть самую выгодную), она представлена на рисунке 1. Как мы видим, она не очень похожа на тРНК. Затем я повысила процент, на который энергия полученной структуры может отклоняться от оптимальной (с 5% до 20%), получила 5 различных структур. Третья из полученных напоминает тРНК больше всех, она представлена на рисунке 2.
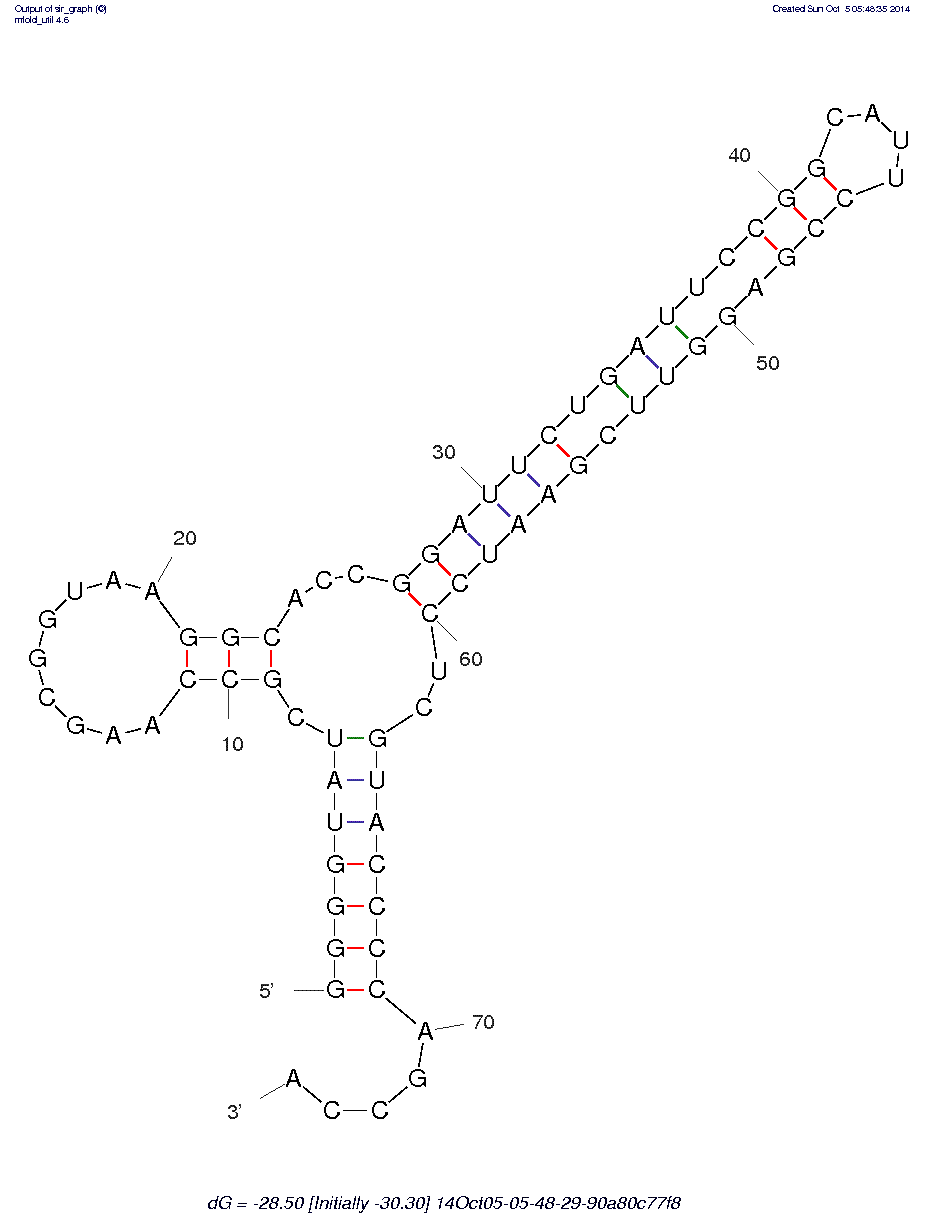
Рис. 1. Наиболее оптимальная структура РНК, полученная с помощью алгоритма Зукера.
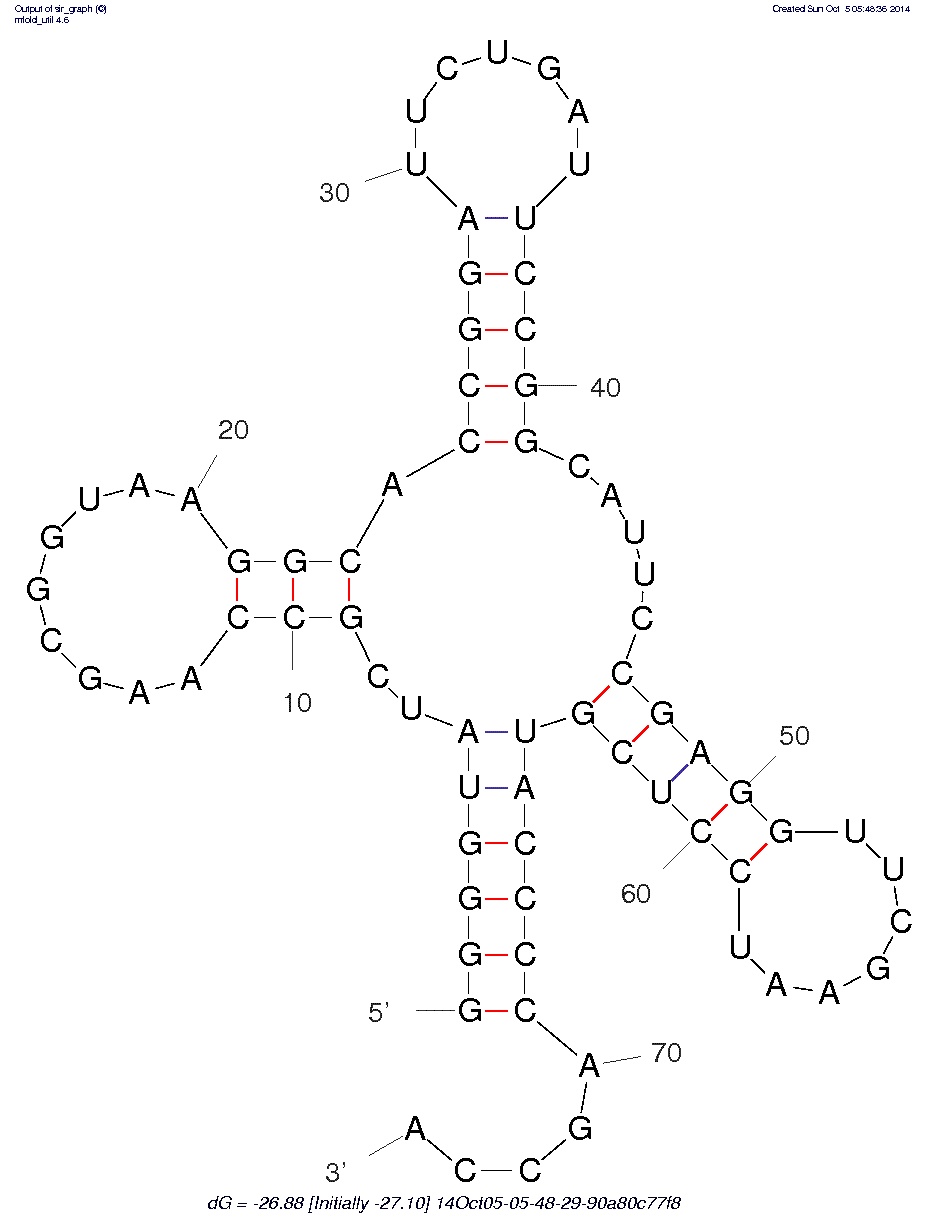
Рис. 2. Похожая на тРНК структура, полученная с помощью алгоритма Зукера.
Теперь вспомним, что тРНК во всех случаях имеет вполне определённую структуру. Номера остатков, которые образуют стебли, одинаковы для всех тРНК. На рисунке 3 представлена типичная структура тРНК (картинка взята из гугла). Как мы видим, остатки 1-7:66-72 образуют акцепторный стебель, 10-13:22-25 - D-стебель (дигидроуридиновый стебель), 27-31:39-43 - антикодоновый стебель, 49-52:62-65 - Т-стебель (псевдоурациловый стебель).

Рис. 3. Типичная структура тРНК.
Составим таблицу по методам, которые мы использовали, чтобы предсказать структуру тРНК, чтобы сравнить их эффективность.
Таблица 1. Сравнение программ find_pair, einverted и mfold.
| Участок структуры | Позиции в структуре (по результатам find_pair) | Результаты предсказания с помощью einverted | Результаты предсказания по алгоритму Зукера |
| Акцепторный стебель | 2-7:66-71 (6 пар) | 1-6:64-69 (6 пар) | 1-6:64-69 (6 пар) |
| D-стебель | 10-12:23-25 | 10-13:53-56 (он пытался) | 9-11:21-23 |
| T-стебель | 49-52:62-65 | - | 49-53:59-63 |
| Антикодоновый стебель | 26-30:40-44 (5 пар) | - | 25-29:39-43 |
| Общее число канонических пар нуклеотидов | 19 | 24 | 19 |
Поиск ДНК-белковых контактов в заданной структуре
Мне был выдан белок с pdb-идентификатором 1PP8 - это белок связывания инициатора транскрипции. Оценим его контакты с ДНК при помощи Jmol. Для этого был написан скрипт, который задаёт множества атомов ДНК и множества атомов белка. Будем считать полярным контактом белка и ДНК ситуацию, когда полярный атом на ДНК контактирует с полярным атомом белка, а неполярным контактном - неполярный атом ДНК с неполярным атомом белка. Полярными будем считать азот и кислород, неполярными - углерод, фосфор, серу. Результаты такого поиска контактов приведены в таблице 2.
Таблица 2. Поиск контактов ДНК-белковых контактов с помощью Jmol (результаты дле всего белка, а не только лдля цепей M, R, Y)
| Контакты атомов белка с | Полярные | Неполярные | Всего |
| Остатками 2'-дезоксирибозы | 10 | 5 | 15 |
| Остатками фосфорной кислоты | 57 | 15 | 72 |
| Остатками азотистых оснований со стороны большой бороздки | 8 | 3 | 11 |
| Остатками азотистых оснований со стороны малой бороздки | 5 | 10 | 15 |
Результаты кажутся мне странными, поскольку по идее белок должен узнавать основания, а не взаимодействовать с сахарофосфатным остовом. С другой стороны - контактов с малой бороздкой больше, чем с большой - так и должно быть, т.к. в основном ферменты взаимодействуют с малой бороздкой ДНК.
Теперь оценим ДНК-белковые контакты с помощью программы nucplot на kodomo. Для этого я сначала перевела свой PDB файл в старый формат с помощью программы remediator, затем запустила nucplot. Однако на полном файле программа не сработала так, как надо, поэтому я вырезала из исходного PDB файла только те цепи, с которыми мне нужно работать (M, R, Y), оставила шапку и информацию про гетероатомы. Получился новый PDB файл, который я также перевела в старый формат и запустила nucplot.
Скачать исходный PDB файл. Скачать сделанный мной PDB файл. Скачать файл в старом PDB формате, с которой работала nucplot. Скачать PS файл, сгенерированный nucplot.
Схему ДНК-белковых контактов можно посмотреть на рисунке ниже.
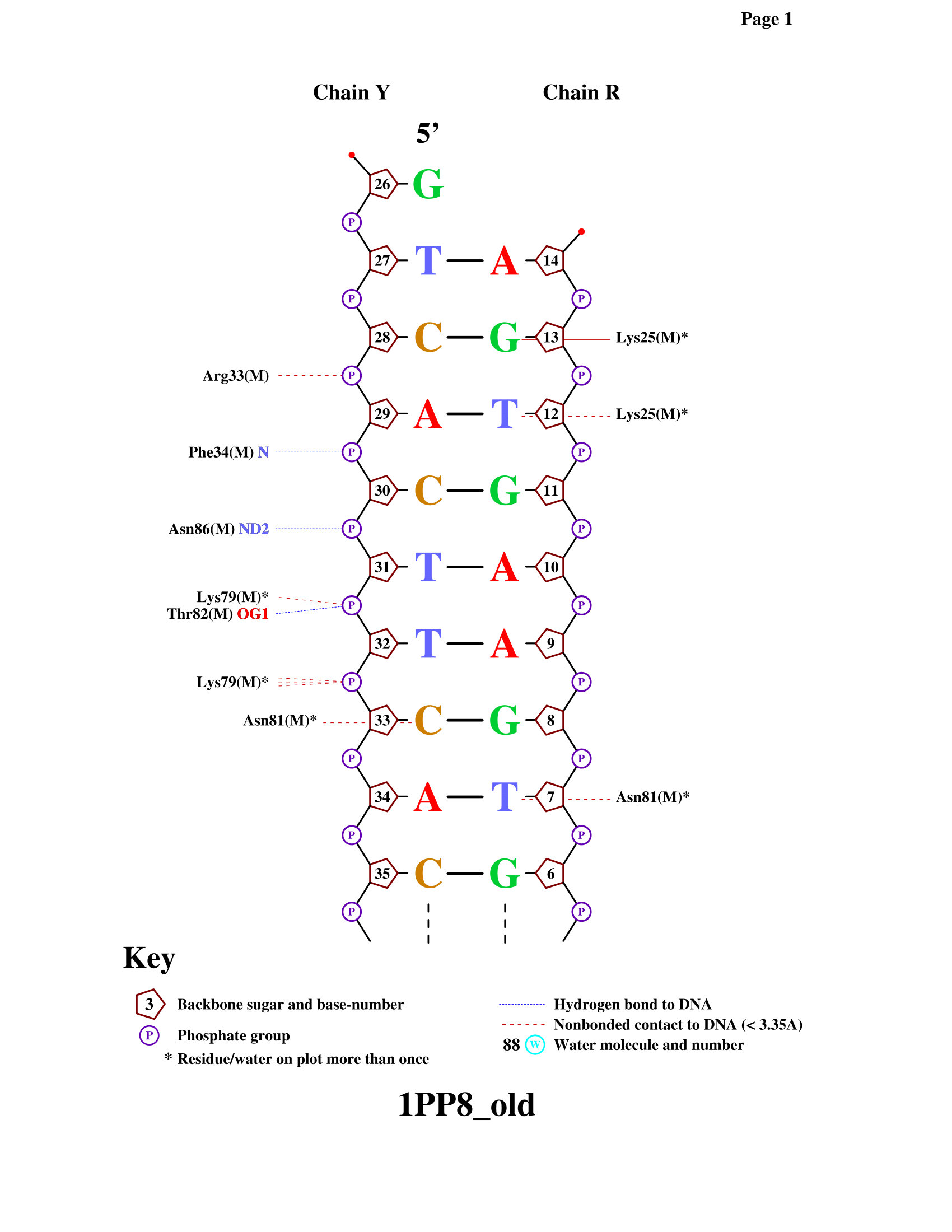
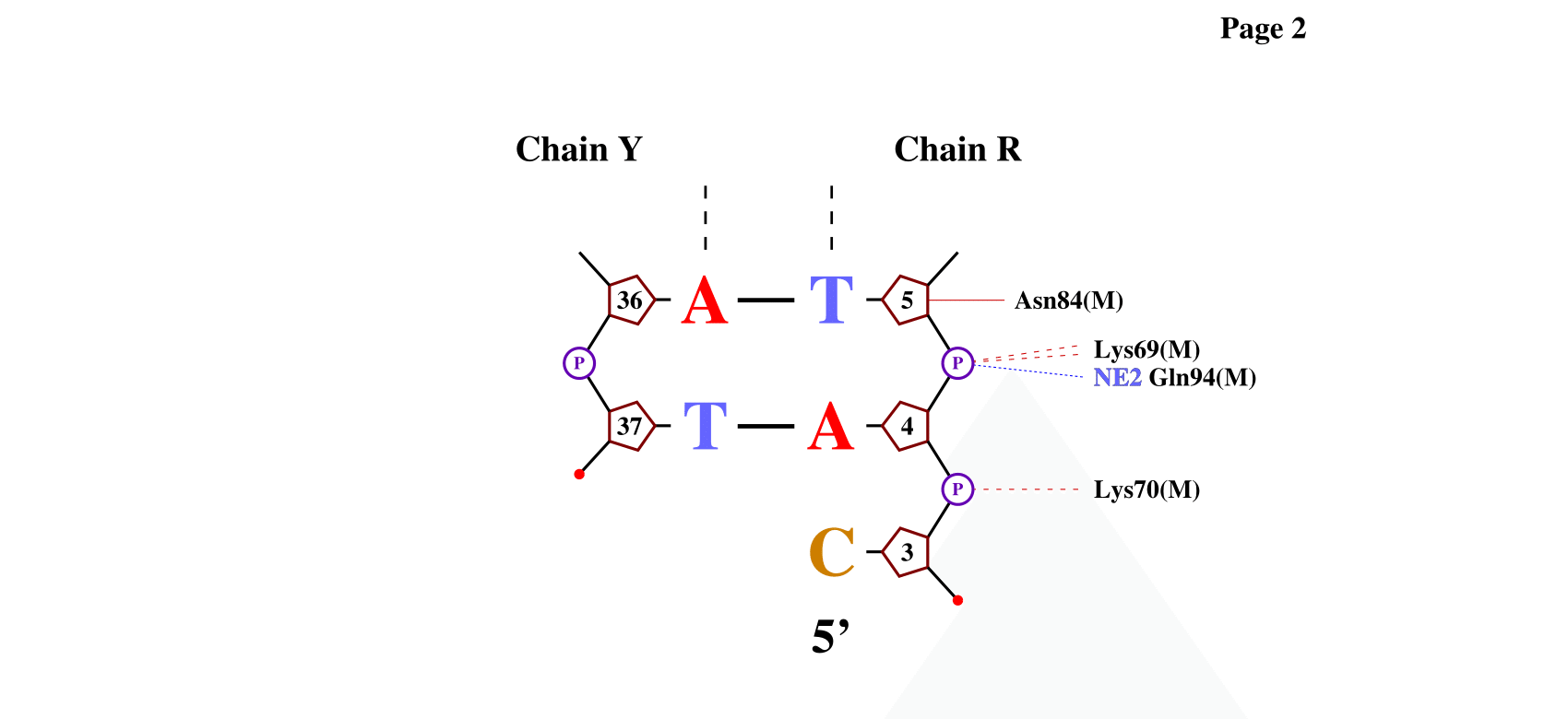

Рис.4. Схема ДНК-белковых контактов для цепей M, R, Y из PDB файла 1PP8, полученная с помощью программы nucplot.
По результатам работы программы видно, что аспарагин 81, лизин 79 и лизин 25 контактируют с белком по два раза каждый. Однако я считаю, что остаток аспарагин 81 важнее, так как он взаимодействует с основаниями, в то время как другие отстатки взаимодействуют с сахарофосфатным остовом. На рисунке 5 можно посмотреть, как выглядит взаимодействие атомов аспарагина и оснований ДНК. Это взаимодействие - полярное (между азотом и кислородом), по сути это водородные связи.
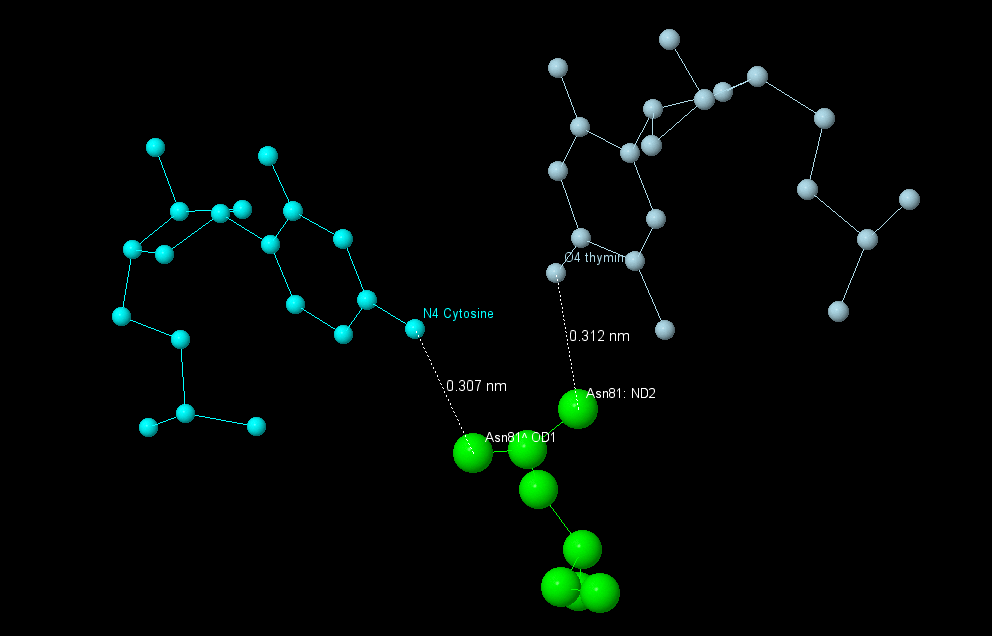
Рис.5. Схема взаимодействия аспарагина, цитозина и тимина, полученная с помощью Jmol.