ФББ 2013-2014


ФББ 2013-2014
EMBOSS
В задании 1 требовалось найти открытые рамки считывания в файле EMBL. Для начала скачаем этот файл с помощью команды entret embl:D89965. Полученный файл можно скачать по ссылке.
Содержимое данного файла - нуклеотидная последовательность мРНК из Rattus norvegicus. Белок, кодируемый этой мРНК связан с серотониновым рецептором крысы, секретируется в желудке. Также в EMBL файле даны ссылки на файлы других баз данных, которые имеют отношение к этому гену. Заглянем в некоторые из них. Например, проверим PDB-файлы, которые по аннотации вроде бы должны содержать в себе 3Д структуру белка. Если начать проверять эти файлы, то окажется, что большинство из них не имеют никакого отношения не то, что к серотониновым рецепторам, но даже к крысам. Например, под PDB идентификатором 1HQY лежит АТФаза из E.coli, а 4G4E - протеасома. Возможной причиной этого может быть то, что произошли ошибки при аннотации, поскольку она происходит автоматически. Также возможно, что на самом деле была расшифрована не последовательность крысы, а последовательность какой-нибудь бактерии, которая обитает внутри неё. Хотя последняя версия не объясняет, почему в EMBL файле так много ссылок на совершенно разные PDB структуры.
Теперь получим набор всех трансляций открытых рамок считывания с помощью команды getorf. Сначала запустим getorf -auto D89965.entret, с автоматическими параметрами. Однако в получившемся файле получается слишком много вариантов, многие из которых, очевидно, не имеют смысла.
Теперь воспользуемся опциями программы getorf. Опция -table 0 говорит программе, что нужно пользоваться только стандартной таблицей генетического кода. Зададим минимально возможный размер опцией -minsize 60. Наконец, скажем программе начинать трансляцию только со старт-кодона, а заканчивать только стоп-кодоном с помощью -find 1. Итоговая команда будет выглядеть так:
getorf -table 0 -minsize 60 -find 1
Полученный файл можно найти здесь. Как видно, результатов получилось намного меньше. Чтобы понять, какая из рамок считывания оказалась правильной, вернёмся к файлу EMBL. В нём записана последовательность белкового продукта данного гена, а именно:
FT /protein_id="BAA14040.1" FT /translation="MALMHFQFTFKQFEQRKSIRSTARKARDDFVVVQTADLFHVAFHY FT GIAQRGLTITSDDHMAVTAYAYYSCHELTPWLRIQSTNPVQKYGA"
В файле, полученным с помощью программы getorf, есть вариант такой же трансляции:
>D89965_4 [163 - 432] Rattus norvegicus mRNA for RSS, complete cds. MALMHFQFTFKQFEQRKSIRSTARKARDDFVVVQTADLFHVAFHYGIAQRGLTITSDDHM AVTAYAYYSCHELTPWLRIQSTNPVQKYGA
Данная запись EMBL ссылается на запись swiss-prot с идентификатором P0A7B8. Получим её с помощью команды entret sw:P0A7B8. Файл с записью можно скачать здесь. Теперь сравним белковую последовательность, которая записана в этом файле с тем, что было ранее. Трансляция из этого файла выглядит так:
SQ SEQUENCE 176 AA; 19093 MW; 3B35E01F51486965 CRC64;
MTTIVSVRRN GHVVIAGDGQ ATLGNTVMKG NVKKVRRLYN DKVIAGFAGG TADAFTLFEL
FERKLEMHQG HLVKAAVELA KDWRTDRMLR KLEALLAVAD ETASLIITGN GDVVQPENDL
IAIGSGGPYA QAAARALLEN TELSAREIAE KALDIAGDIC IYTNHFHTIE ELSYKA
Она не похожа ни на одну рамку считывания, полученную с помощью getorf и на трансляцию из файла EMBL. Также стоит отметить, что в файле Swiss-Prot стоят ссылки на те же PDB файлы, что и в записи EMBL, однако по описанию в поле CC запись аннотирована как протеасома. Как уже было указано ранее, скорее всего это связано с тем, что этот белок был получен не из клетки крысы, а из клетки E.coli, которая жила в этой крысе.
Файлы-списки
Теперь получим файл-список всех алкогольдегидрогеназ. Их идентификаторы описываются записью adh*_*. Сначала я попробовала воспользоваться программой entret, введя команду entret sw:adh*_* adh.fasta. В итоге получился файл, в котором действительно содержалась информация о всех алгокольдегидрогеназах человека, однако это были их полные swiss-prot записи. Стало очевидным, что надо использовать программу, которая выдаст только последовательности данных белков.
Используем команду seqret sw:adh*_* adh.fasta. Теперь мы получили нужный файл со всеми последовательностями алькогольдегидрогеназ в формате fasta.
Теперь получим файл, содержащий только их universal adress (USA). Для этого воспользуемся программой infoseq с опциями -only -usa. Итоговая команда запишется как infoseq -only -usa -outfile USA sw:adh*_*. В итоге получили файл с нужным нам списком всех USA.
Затем необходимо было сделать список из алкогольдегидрогеназ определённых видов. Мне достались банальные человек (HUMAN), курица (CHICK) и дрозофилы (DROPS, DROAR), метаногенная анаэробная архея Methanococcus maripaludis (METM5), экстремофильная галофильная архея Halobacterium salinarum (HALSA).
USA последовательностей данных организмов были выделены в отдельный файл с помощью команды grep -f spec.txt USA > USA_mini, где spec.txt - это список идентификаторов "моих" видов. В итоге получился этот файл.
Чтобы подать командам EMBOSS на вход файл-список необходимо поставить значок @ перед этим самым файлом. Отфильтруем последовательности алкогольдегидрогеназ, чтобы получить файлы для белков только из "моих" организмов. Для этого воспользуемся командой seqret @USA_mini adh.fasta, на выходе получим файл с отфильтрованными последовательностями, который можно скачать здесь.
Случайная модель для оценки достоверности выравнивания
Выберем две алкогольдегидрогеназы из предыдущего задания, пусть это будут ADH_CHICK из Gallus gallus и ADH_METM5 из Methanococcus maripaludis. Филогенетически эти организмы очень далеки друг от друга, поэтому скорее всего их белки окажутся негомологичными и будут иметь маленькое сходство в последовательностях. Построим их локальное парное выравнивание с помощью команды "water". Команда будет выглядеть так: water sw:ADH*_METM5 sw:ADH*_CHICK. Вес получившегося выравнивания - 48.0.Полученное парное выравнивание можно скачать здесь. Теперь перемешаем вторую последовательность (из курицы) 100 раз с помощью программы shuffleseq. Команда будет выглядеть так: shuffleseq sw:ADH*_CHICK -shuffle 100 CHICK_messedup.fasta.
Построим локальное парное выравнивание с помощью water 100 получившихся последовательностей с исходной ADH_METM5 (полученный файл - здесь). Получим 100 парных выраваний, у каждого из которых будет свой вес. С помощью команды grep "Score" coolstory.fasta > scores.txt получим список весов всех выраниваний (файл с весами - тут).
По получившимся весам построим гистограмму в Excel, предварительно упорядочив веса по возрастанию. По оси Х откладываем номер, по оси У - сам вес. Получившаяся гистограмма представлена на рисунке 1. На ней стрелкой показано место веса выравнивания настоящих белков. Очевидно, что белки негомологичны, так как вес выравниваний некоторых случайных последовательностей оказывается намного выше, чем вес оригинального выравнивания. Score - это параметр, по которому нельзя однозначно сказать, что белки гомологичны или нет, всегда нужно рассматривать его в совокупности с другими параметрами, например e-value или score других выравниваний.
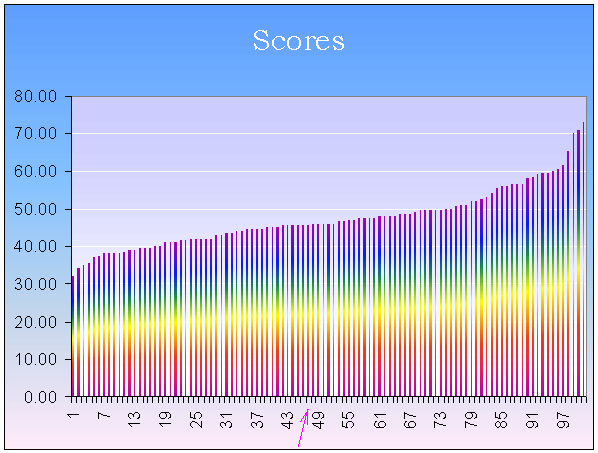
Рис.1Распределение весов выравниваний 100 случайных последовательной с ADH_METM5 и 1 настоящей. Стрелкой показано положение веса настоящего выравнивания (48.0)