ФББ 2013-2014


ФББ 2013-2014
Standalone BLAST
Поиск некодирующих РНК, аннотированных в одном штамме, в геноме другого штамма
Скачаем с FTP сервера NCBI файл, который содержит все РНК из организма Bacillus subtilis штамма 168 uid57675. Типы РНК, содержащиеся в файле - транспортная РНК, рибосомальная РНК, так называемая miscellaneous_RNA (misc_RNA) - это некодирующие РНК (т.е. РНК, которые не транслируются в белки). Всего в данном файле содержится 63 последовательности misc_RNA.
Сначала с помощью команды grep отберём только информацию о misc_RNA. Команда: "infoseq "NC_000964.frn" | grep "misc_RNA" > misc_RNA.fa". В полученном файле вручную удалим все столбцы, кроме первого (список USA). Чтобы получить список всех misc_RNA вместе с их последовательностью, применим следующую команду: "seqret @USA.fa misc_RNA_seq.fa".
Файлы, полученные в процессе работы:
Далее было необходимо скачать аналогичные файлы для организма Bacillus subtilis spizizenii TU B 10 uid73967, с расширением .gbk, с расширением .fna.
По .fna файлу для нового штамма составим базу данных с помощью программы: "makeblastdb -in NC_016047.fna -dbtype nucl" (программа взята из подсказок лдля Standalone BLAST). В результате работы этой программы были получены файл 1, файл 2, файл 3, в них и содержится база данных.
Теперь запустим blastn, чтобы получить нужные нам выравнивания. Программа выглядит так: "blastn -task blastn -query misc_RNA_seq.fa -db NC_016047.fna -outfmt 7 -num_alignments 1 -out alignment.fa" (программа взята из тех же подсказок). Получился файл c выравниваниями последовательностей misc_RNA из первого штамма Bacillus subtilis с последовательностями их гомологов из второго штамма той же бактерии.
В результате была получена таблица Excel, где суммированы данные о выравнивании, гомологе и изначальной полседовательности. Её можно скачать здесь (лист 1) или посмотреть на рисунке 1.

Рис.1. Итоговая таблица Excel по всем misc_RNA Bacillus subtilis
Поиск гомологов РНК Bacillus subtilis в геноме другой бактерии
С того же FTP сервера NCBI был скачан геном бактерии Bacillus cereus FRI 35 uid173403. Его будем использовать как базу данных (по аналогии со вторым штаммом в первом задании). Проведём поиск гомологов тремя способами:
По данным этих выравниваний была составлена таблица Excel (лист 2), в которой подсчитано количество найденных гомологов с e-value <0,001. Увидеть её можно на рисунке 2.


Рис.2. Cравнение результатов выравнивания, полученных с помощью различных алгоритмов.
Очевидно, что megablast ищет самые близкие (а потому похожие) гомологи исходного гена, поэтому находок так мало, но зато мы более или менее уверены в их достоверности. Blastn со стандартными параметрами находит меньше, чем Blastn с изменёнными. Две эти программы могут обнаружить более отдалённый гомолог заданной последовательности, который по критериям megablast не подойдёт. Как видно из заданий 1 и 2, megablast удобно применять на очень близких организмах (например, штамм одного и того же вида), так как находок всё равно будет не так много и можно будет утверждать, что они достаточно достоверны. Но на более далёких организмах (например, разные виды одного и того же рода), megablast уже может не подойти для поиска гомологов, и тогда надо будет воспользоваться программами с менее строгими критериями.
Поиск неправильно аннотированных генов программой blastx
Скачиваем с FTP сервера NCBI файл, в котором лежат все белки Bacillus subtilis spizizenii TU B 10 uid73967. Теперь он будет базой для поиска гомологов misc_RNA из первого штамма Bacillus subtilis. Предполагается, что гомологов обнаружиться не должно, поскольку misc_RNA - это РНК, а в качестве базы мы используем нечто белковое.
Команда для создания базы: "makeblastdb -in NC_016047.faa -dbtype prot".
Команда для выравнивания: "blastx -query misc_RNA_seq.fa -db NC_016047.faa -out wrongway.fa -outfmt 7 -evalue 0.0001"
Нашлось несколько белковых "гомологов" к изначальным РНК. Они представлены на рисунке 3.
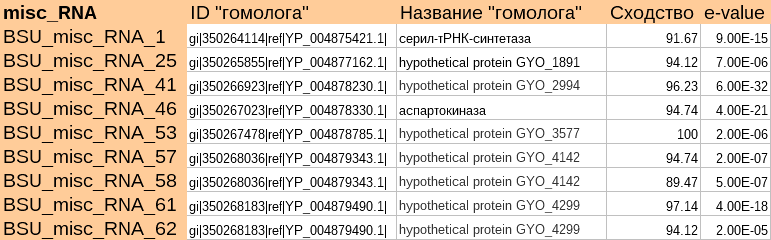
Рис.3. Найденные белки-"гомологи" изначальных misc_RNA и данные по выравниваниям
Как видно из таблицы, ошибочно были найдены не только гипотетические белки, но и 2 подтверждённых белка - серил-тРНК синтетаза и аспартокиназа. Если запустить blastp для серил-тРНК синтетазы, то будут найдены консервативные домены, а также много гомологов с хорошим сходством и e-value, что может свидетельствовать о достоверности этого варианта аннотации. Для аспартокиназы - аннотация точно ошибочная, поскольку под идентификатором Refseq лежит очень короткая последовательность этого белка.