2013-2014


2013-2014
Реконструкция филогении
Используестя тот же список бактерий, для которых строилось дерево в предыдущем практикуме. Филогения будет восстановлена по белку - фактору элонгации G. Этот фактор, как и другие белки, участвующие в транскрипции, является консервативным. Он осуществияет ГТФ-зависимую транслокацию рибосомы, отвечая за освобождение Р-участка тРНК и перемещение туда продукта элонгации.
Поисковый запрос в UNIPROT выглядел следующим образом:
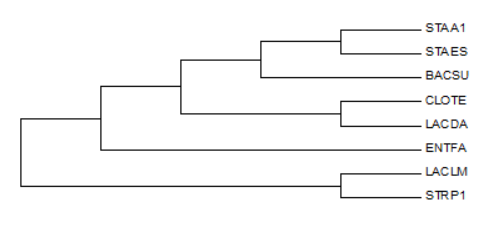
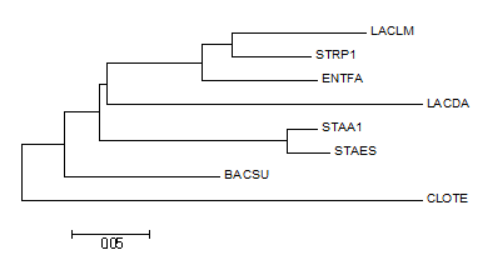
EFG_BACSU, EFG_CLOTE, EFG_ENTFA, EFG_LACLM, EFG_STRP1, EFG_STAA1, EFG_STAES, EFG_LACDAПолучили файл, в котором лежат последовательности всех нужных белков. Далее с помощью сервиса Uniprot было выполнено выравнивание этих последовательностей. Результат в формате .fasta можно скачать здесь. На рис. 1 показано дерево, построенное в Uniprot автоматически. На рис.2 показан участок выравнивания белков, открытый в Jalview (одинаковые для всех белков аминокислоты выделены синим).

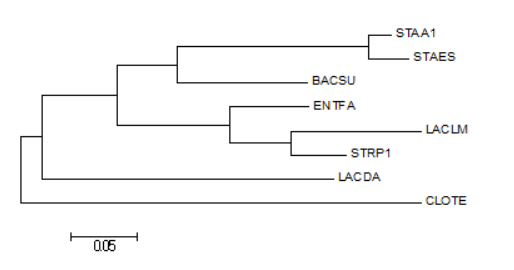
Рис.1 Филогенетическое дерево, построенное на основании выравнивания белковых последовательностей факторов элонгации G (автоматически с помощью Uniprot)
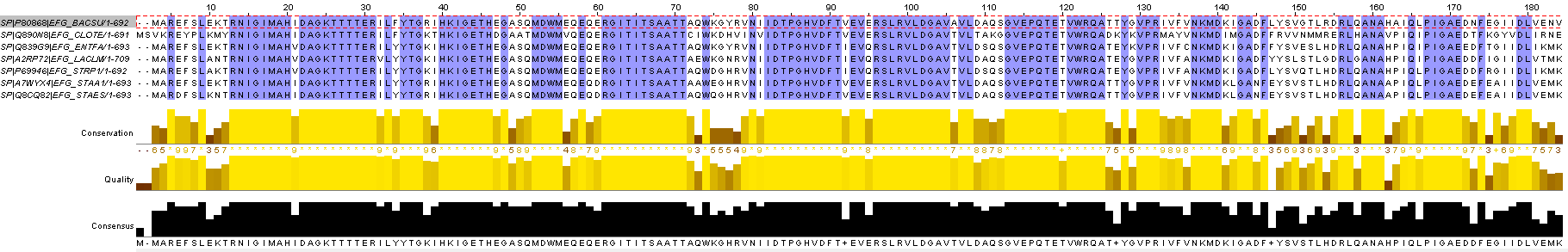
Рис.2 Участок выравнивания, открытый в Jalview. Наиболее консервативные позиции (100% совпадение) выделены синим.
Теперь создадим собственный Jalview-проект, где в окне с выравниваниями будет только мнемоника вида, а дерево будет построено с помощью алгоритма Neighbor Joining Using % Identity. Выравнивание аналогично рис. 2, получившееся дерево представлено на рис.3. Как видно, филогенетическое дерево совпадает с деревом, построенным мной в предыдущеем практикуме на основе данных NCBI Taxonomy.
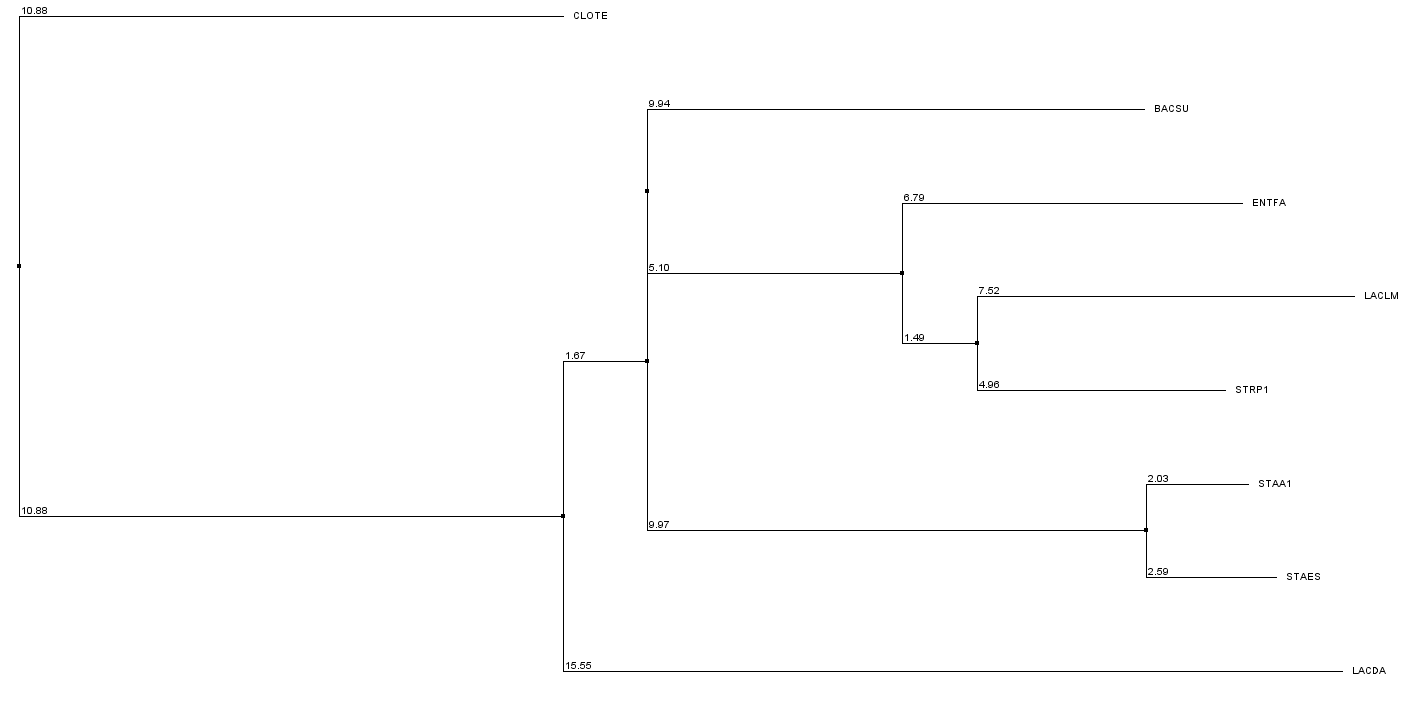
Рис.3. Филогенетическое дерево, построенное в Jalview по алгоритму Neighbor Joining Using % Identity на основе поданного на вход выравнивания.
Это дерево можно сохранить в Newick формате: ((LACDA:15.550489,((STAES:2.5946722,STAA1:2.0336618):9.97107,(((STRP1:4.957924,LACLM:7.524544):1.4857979,ENTFA:6.789097):5.1046057,BACSU:9.93747):0.0):1.6698818):10.878331,CLOTE:10.878331); скачать этот файл можно по ссылке.
Теперь откроем выравнивание в программе MEGA и с помощью программы Phylogeny построим филогенетические деревья, используя разные алгоритмы.
Алгоритм Maximum likehood
Алгоритм Maximum likehood
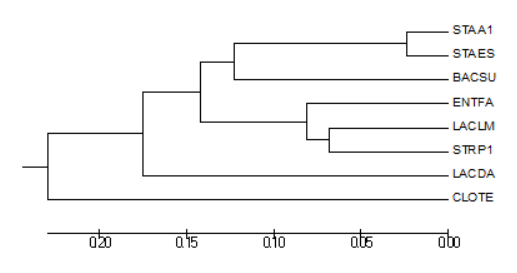
Алгоритм Minimum-evolution

Алгоритм UPGMA
Алгоритм Maximum pairsmony