Mini-review of Bacteroides thetaiotaomicron genome:
protein length distribution and codon frequencies
protein length distribution and codon frequencies
учебная страница панькиной вари
| Taxonomy rank | Name |
| Domain | Bacteria |
| Kingdom | Pseudomonadati |
| Phylum | Bacteroidota |
| Class | Bacteroidia |
| Order | Bacteroidales |
| Family | Bacteroidaceae |
| Species | Bacteroides thetaiotaomicron |
Bacteroides thetaiotaomicron is a typical representative of the genus Bacteroides. It is known for its role in humans and other mammals' intestinal microbiota [2].
Bacteroides thetaiotaomicron is notable for its role in the digestion of various complex polysaccharides. This species is one of the main gut symbionts, making its study significant in the context of research on symbiotic relationships, gastrointestinal health, and the development of the immune system [3]. The ability to utilize various polysaccharides is ensured, in part, by the presence of clusters of termed polysaccharide utilisation loci (PULs) [4].
In this mini-review, we analyze the Bacteroides thetaiotaomicron genome. We investigated the coding regions composition of present replicones and several coding regions features, such as protein length and codon frequencies.
We used genome feature tables and coding sequence files obtained from the NCBI database (1). In the first part of the analysis, we used Google Sheets(3) to examine the distribution of coding sequences across the bacterial replicons. With data from the file GCF_000011065.1_ASM1106v1_cds_from_genomic.fna (2), we generated a histogram of protein lengths. To analyze codon frequencies, we used data from the mentioned file(2) and processed them with Python(4). We estimated the difference in synonyms codon frequencies in identified and hypothetical proteins with two-proportion z-test:
We examined the number of protein coding genes and different RNAs in both present replicones from the genome(1). The results are summarized in Table 1. In the genome of Bacteroides thetaiotaomicron, two replicons are present: one circular chromosome with a length of 6.26 Mb and one circular plasmid with a length of 33 kb. The majority of coding sequences in the main chromosome are protein-coding genes — 4,649 in total; additionally, it contains 70 tRNA genes and 15 ribosomal RNA genes. The second replicon, the plasmid p5482, contains 38 protein-coding genes, whose primary function is environmental sensing [5].
| Protein coding genes | tRNA | rRNA | tmRNA | |
| Chromosome | 4,649 | 70 | 15 | 1 |
| p5482 plasmid | 38 | 0 | 0 | 0 |
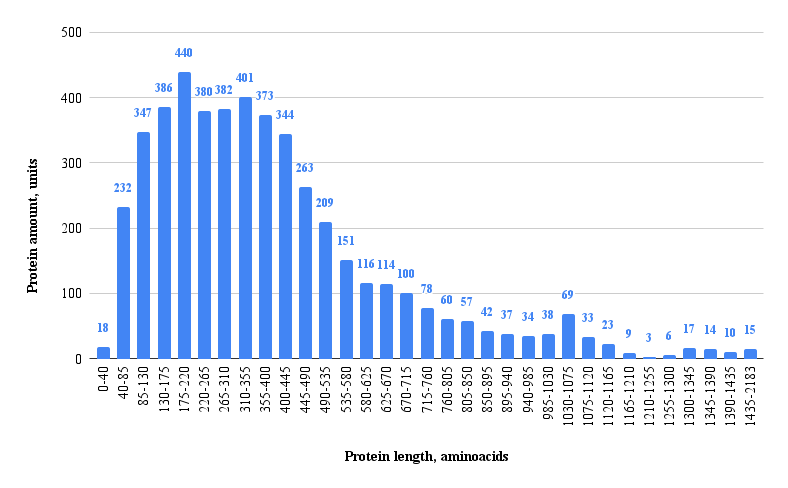
Analysis of the protein length distribution within the Bacteroides thetaiotaomicron genome (Figure 1) reveals a characteristic right-skewed profile, indicative of a high abundance of shorter polypeptides and a rapid decline in frequency with increasing length. The total amount of analyzed proteins is 4801 units. The distribution is unimodal, with a pronounced peak observed in the shortest length groups, where the number of proteins reaches a maximum of 440 proteins per bin. In particular, we can note that the peaks in protein abundance are located around the median value of 390 amino acids. It corresponds to our data in google sheets (2). The data demonstrates a long-tailed distribution, with a substantial number of length bins containing proteins, albeit at low frequencies (e.g., less then 50 proteins per bin beyond the mid-range), confirming the presence of a limited set of elongated proteins. This overall pattern, dominated by a high prevalence of short proteins, is consistent with genomic characteristics commonly observed in prokaryotic organisms [6].
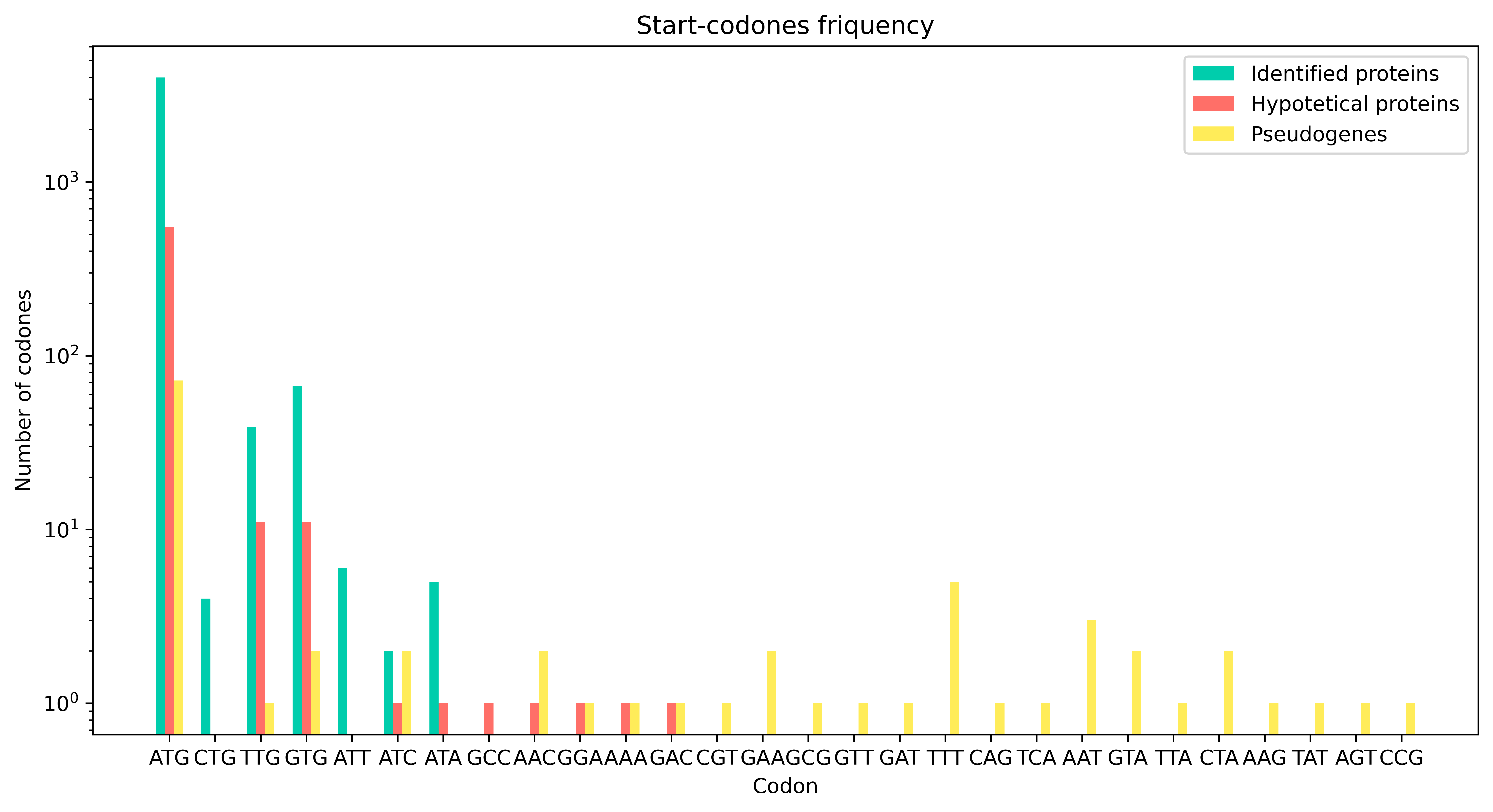
We examined codon frequencies in bacterial coding sequences. We hypothesized that codon frequencies would differ across different groups of CDSs. Therefore, we divided them into three groups according to its description in CDS table (2): identified proteins — proteins with defined names in cds table (2) (4117 sequences), hypothetical proteins – defined as proteins without details (577 sequences), and pseudogenes (107 sequences).
This separation arises because codon usage in protein-coding sequences is subject to selective pressure, as codon frequencies constitute one of the mechanisms regulating translation [7]. In contrast, codons in pseudogene sequences are no longer constrained by selection due to the loss of their expression [8].
Hypothetical proteins were also separated from the set of identified proteins, as no reference sequences are available for them. Consequently, it is not possible to reliably determine whether they contain insertions such as mobile genetic elements or self-splicing introns, which present in numerous bacterial genomes [9].
We identified the first codon (the first three nucleotides) of the identified proteins, hypothetical and pseudogene sequences. According to Figure 2 , the dominant start codon is the canonical methionine codon AUG. Both identified and hypothetical sequences also contain alternative start codons — CTG and TTG encode leucine, GTG encodes valine, and ATT, ATC, and ATA encode isoleucine.
In pseudogenes, however, we detected an additional 21 codons (5 of which also appear in genomic sequences) that are not known to function as start codons.
These ‘novel start codons’ in pseudogenes are most likely the result of accumulated neutral mutations [8]. The occurrence of various amino-acid-encoding codons at putative start positions in genomic proteins suggests that some of these sequences may also be pseudogenes, as pseudogenes are often misannotated as hypothetical proteins during initial genome annotation [10].
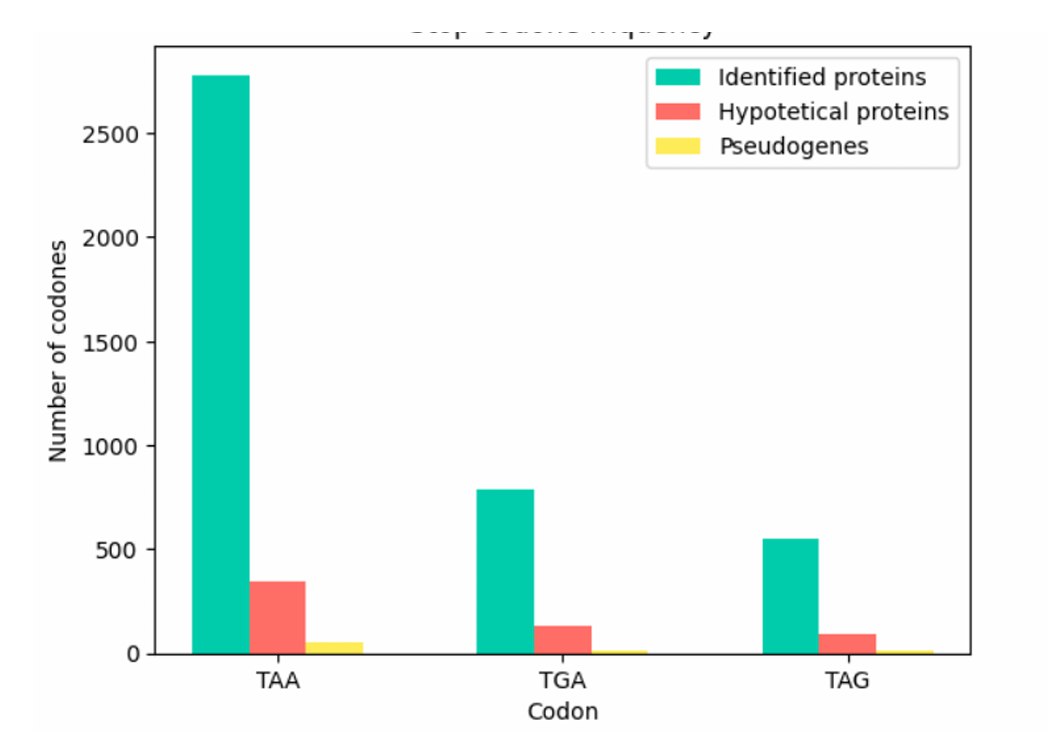
We identified the terminal three nucleotides (stop codons) of the identified proteins, hypothetical proteins and pseudogene sequences. In both identified and hypothetical one sequences, only the canonical stop codons TAA, TGA, and TAG were observed, with TAA occurring approximately five times more frequently than TGA or TAG (Figure 3).
Pseudogenes also predominantly use these three stop codons, but we additionally detected 23 codons that encode amino acids. This likely reflects the mutational decay of pseudogenes, which can obscure their true stop codons [8].
We compared the frequencies of synonymous codons between the identified and hypothetical protein sets. This analysis revealed significant differences in codon usage for 10 amino acid codons , z-criterion > 2.59 (Table 3T; full dataset in (5)). The data further show that hypothetical proteins exhibit a weaker codon bias than identified proteins, with codon usage appearing more stochastic.
Based on these observations, we propose hypothetical proteins do not represent a single category of uncharacterized proteins but rather a heterogeneous set that likely includes horizontally acquired genes, pseudogenes, and genuinely functional proteins. These subgroups, and how they can be distinguished, are discussed in detail in the following section.
| AA | Codon | Identified (%) | Hypothetical (%) | Z-criterion |
| A | GCC | 27.645 | 22.715 | 2.497 |
| D | GAC | 40.928 | 34.313 | 3.0362 |
| D | GAT | 59.072 | 65.687 | -3.0362 |
| F | TTC | 51.085 | 42.28 | 3.9616 |
| F | TTT | 48.915 | 57.72 | -3.9616 |
| G | GGG | 9.936 | 12.739 | -2.0762 |
| I | ATC | 39.171 | 28.64 | 4.8839 |
| I | ATA | 22.884 | 32.536 | -5.079 |
| L | CTG | 32.142 | 23.993 | 3.9588 |
| L | CTA | 4.93 | 7.071 | -2.1706 |
Our analysis generated a set of bioinformatic characteristics for one of the sequenced B. thetaiotaomicron genome, which can be applied to several practical tasks. In particular, frequencies of start, stop, and synonymous codons can help prioritize hypothetical proteins for further study. Hypothetical proteins that use canonical start and stop codons and display codon preferences similar to identified proteins are more likely to be functional.
For such candidates, calculating the Codon Adaptation Index (CAI)[12] may further indicate how closely they resemble known functional proteins. Conversely, sequences that diverge from these patterns are likely pseudogenes, potentially recently formed if they still retain features that led to their initial annotation as proteins[10].
Distinct codon biases may also indicate horizontal gene transfer, which can be assessed by comparing codon usage and GC content with other predominant species of the mammalian gut microbiome [13].