Сборка de novo
Петренко Павел
Факультет биоинженерии и биоинформатики, Московский Государственный Университет имени М.В.Ломоносова
Введение
В этом практикуме попробуем собрать геном бактерии Buchnera aphidicola str. Tuc7 из коротких ридов длиной 39 нуклеотидов, полученных с помощью Illumina: SRR4240359.
Скачаем архив с чтениями в рабочую директориюс помощью команды wget:
Подготовка чтений программой trimmomatic
Начнём с удаления возможных остатков адаптеров, чтобы оставить только качественные фрагменты в ридах. Сначала объединим их все адаптеры в один файл:
Посчитаем количество отобранных адаптеров (всего получилось 27):
Наконец-то удалим адаптеры из ридов:
Увидели, что у нас удалилось 55872 рида, что составляет 0.41% от общего количества:
Теперь удалим с правых концов чтений нуклеотиды с качеством ниже 20, и оставим только такие чтения, длина которых не меньше 32 нуклеотидов:
Опять же посмотрим в выдачу и увидим, что теперь у нас отбросилось ещё 1317986 ридов, аж 9.76%:
Ради интереса посмотрим вес наших файлов и сравним их:
Исходный файл весит 445M.
Файл с обрезанными адаптерами весит 443М.
Файл с ридами лучшего качества (без нуклеотидов низкого качества и коротких ридов) весит 385М.
Видим, что после нашей "чистки" размер файла уменьшился на 60М.
Работа с программой velveth
Velveth - это первая часть конвейера Velvet, программы для сборки геномов из коротких ридов. Воспользуемся velveth, чтобы разбить наши риды на k-меры:
Опции:
31 - длина k-мера
short - работаем с одноконцевыми чтениями
fastq.gz — указание формата файла, который подаём на вход
В результате в папке velvet появилось 3 файла:
Log - информация о запуске и ошибках
Roadmaps - бинарный файл, который содержит "карты" прохождения ридов через граф де Брёна
Sequences - нужные нам k-меры
Работа с программой velvetg
Velvetg - это вторая часть конвейера Velvet, velvetg берёт "сырой" граф k-mers от velveth, очищает его от ошибок, использует парную информацию для соединения фрагментов и выдаёт готовые контиги/скаффолды. Запускаем:
В результате в папку velvet появилось 5 файлов:
contigs.fa - fasta-файл, содержащий последовательности контигов длиной более двух слов из velveth
PreGraph, Graph, LastGraph - три последовательных состояния графа де Брёна в процессе работы Velvetg (каждый следующий файл чище предыдущего)
stats.txt - таблица с разной информацией
Посмотрим, сколько у нас получилось контигов:
Видим, что всего получилось 285 контигов.
N50 = 70607 (10.3% от общей длины).
Отберём 3 самых длинных контига и посчитаем их покрытие:
Находки:
>NODE 11 length 125674 cov 44.550949
>NODE 1 length 108447 cov 42.009186
>NODE 14 length 71403 cov 39.411552
Теперь посмотрим, есть ли у нас аномально большие или маленькие находки. Как я понял, мы берём медианный контиг (получается 143) и смотрим его покрытие, а затем сравниваем с ним все остальные. Найдём медианный контиг:
Получили:
Видим, что медиана очень маленькая, поэтому, скорее всего, мы найдём только контиги с аномально большим покрытием:
Как и ожидалось, контигов с аномально маленьким покрытий нет, зато нашлось 52 контига с аномально большим покрытием. Среди таких, например, >NODE_1_length_108447_cov_42.009186 (c покрытием ~ 42.01%, в 7 раз больше медианного) или >NODE_420_length_76_cov_54.263157 (c покрытием ~ 54.26%, в 9 раз больше медианного).
Анализ
Для работы в этом пункте задачи я выбрал рефференсный геном Buchnera aphidicola str. W106 (Myzus persicae) NZ_CP002699.
Контиг 1

| Range | 113933-140272 |
|---|---|
| Score | 19826 |
| Identities | 21463/26651(81%) |
| Gaps | 701/26651(2%) |
Контиг почти полностью совпадает с рефференсом.
Контиг 11
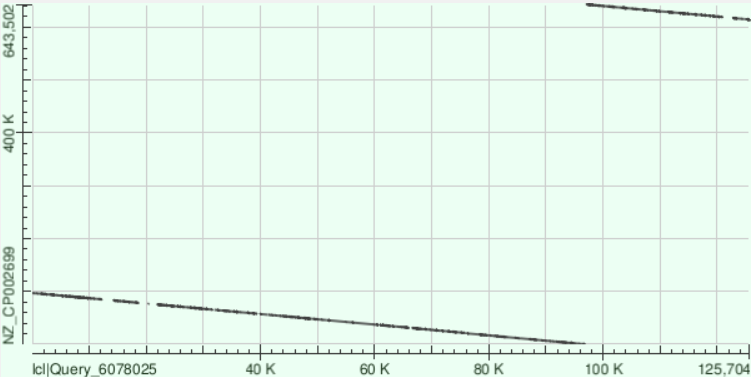
| Range | 628956-643122 |
|---|---|
| Score | 12225 |
| Identities | 11780/14292(82%) |
| Gaps | 272/14292(1%) |
Видим, что на графике есть два хорошо выровнянных участка. Я предполагаю, что такая картина могла образоваться в связи с тем, что у бактерии кольцевая ДНК и начало секвенирования, совпадающее с концом полностью покрылось контигом (сначала захватилась первая область, потом вторая).
Контиг 14
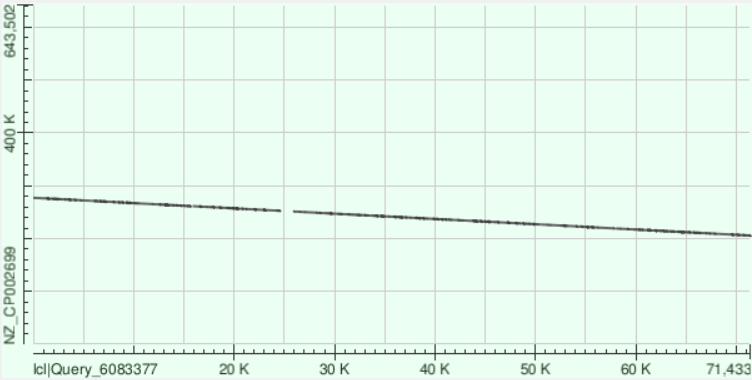
| Range | 223109-235768 |
|---|---|
| Score | 11457 |
| Identities | 10634/12789(83%) |
| Gaps | 240/12789(1%) |
Если сравним график с таковым для первого контига, то увидим, что он наклонён вниз, а не вверх. Это может говорить нам о том, что выравнивание контига пошло по минус-цепи рефференса.