
Из таблицы было выбрано три пары белков из семейства HSP70, принадлежащих представителям разных доменов. С помощью средств bash, последовательности этих белков были извлечены в текстовые файлы. Для определения консервативности последовательностей, отыскания гомологичных белков и выяснеия их родства, с помощью программы Jalview было построено выравнивание 1 с раскраской по схеме ClustalX с условием Identity Threshold = 100%. Из всех предложенных алгоритмов результат работы "Mafft with Defaults" показался мне наиболее правильным. Поля консервативности и др. были скрыты намеренно, чтобы остображалась лишь релевантная информация. Ссылка на программу bash (для вызова в командной строке PuTTY: bash alignment.sh)
Для расчета параметров консервативности была использована команда infoalign из пакета EMBOSS. Команда infoalign и её атрибуты применялись к выравниванию, полученному в программе JalView. Так как в рамках нашего курса гэпом считается несколько подряд идущих символов, для их подсчёта был выбран атрибут -gaps. Если считать количество гэпов в целом выравнивании, то, с помощью программных средств, можно убедиться, что их 207.
| Имя последовательности | Длина последовательности | Длина выравнивания | Абсолютно консервативные позиции | % абсолютно консервативных позиций | Функционально консервативные позиции | % функционально консервативных позиций | Gaps | % gaps | Консервативные на 70% | % консервативных на 70% |
| Clostridium_beijerinckii_DNAK_CLOB8_1-614 | 614 | 739 | 132 | 21,5 | 229 | 30,99 | 20 | 3,26 | 288 | 38,972 |
| Halobacterium_salinarum_DNAK_HALS3_1-629 | 629 | 740 | 132 | 20,99 | 229 | 30,95 | 18 | 2,86 | 288 | 38,919 |
| Methanosaeta_thermophila_DNAK_METTP_1-615 | 615 | 740 | 132 | 21,46 | 229 | 30,95 | 20 | 3,25 | 288 | 38,919 |
| Yersinia_pestis_HSCA_YERPE_1-650 | 650 | 755 | 132 | 20,31 | 229 | 30,33 | 19 | 2,92 | 288 | 38,146 |
| Homo_sapiens_HS71A_HUMAN_1-641 | 641 | 738 | 132 | 20,59 | 229 | 31,03 | 13 | 2,03 | 288 | 39,024 |
| Saccharomyces_cerevisiae_HSP7Q_YEAST_1-657 | 657 | 743 | 132 | 20,09 | 229 | 30,82 | 15 | 2,28 | 288 | 38,762 |

К полученному выравниванию было добавлено поле "Разметка". Оно отражает следующие свойства: G - участки выравнивания, содержащие гэпы; C - участки, консервативные на 80 и более %; F - функционально консервативные участки выравнивания. Видно, что, в общем, в хорошем выравнивании достаточно часто встречаются как консервативные позиции (С), так и абсолютно функционально консервативные позиции (F). На выбранном участке имеется несколько F позиций. Начиная со столбца 530 и заканичвая столбцом 680, было отмечено как минимум по три случая состояния позиций. Так и далее, столбцы с номерами 560 и 581 содержат остатки изолейцина и лейцина: чрезвычайно сходных аминокислот. Позиции 599 и 611 представлены остатками изолейцина и валина. Также можно заметить, что в общее выравнивание содержит восемь крупных блоков, размер которых увеличивается от периферии к центру.
Для имитации эволюционного пути и проверки алгоритмов JalView на способность адекватно отражать такой процесс, было проведено восемь раундов искуственной мутации, эквивалентных 800м годам реального эволюционного процесса для части (1-100) аминокислотной последовательности белка сиалидазы из бактерии Clostridium beijerinckii. Данные манипуляции были проведены с использованием средств bash и пакета EMBOSS. Выбранный для мутирования файл с участком последовательности Clostridium beijerinckii:
>p1 sialidase [Clostridium beijerinckii] MIRRNKRILSLTLSMAVFTTMFMSTSFITKAETVSLGANSEITSNASTESTAVATNIALN KPSTASSVTGGNTASLAVDGNAGTRWESAQGSDPQWISIDВ выравнивании 3 представлены исходная последовательность предполагаемого предка и последовательности восьми раундов мутаций p1> p2 > p3 > ... > p9. При помощи команды msbar каждый раунд вносится семь точечных мутаций различных типов. Решено проводить восемь раундов с целью более вероятного обнаружения ошибки программы в дальнейшем.
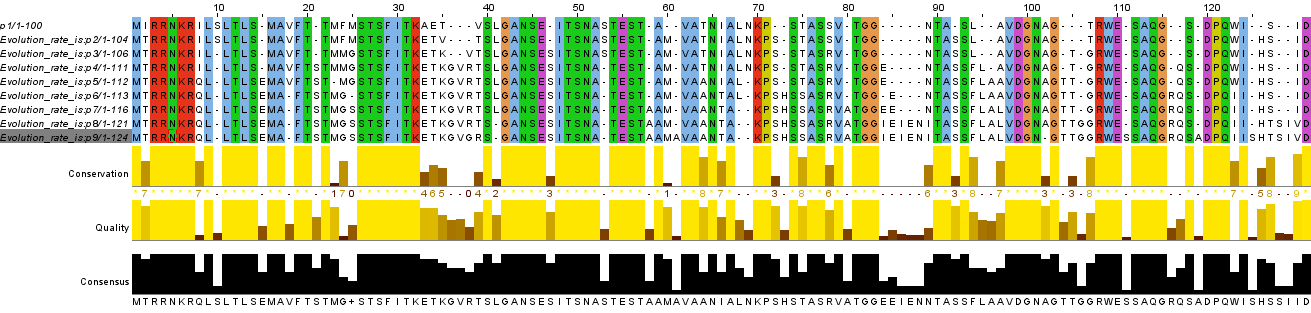

По исправленному выравниванию 4 можно проследить эволюционный путь последовательности:
Для имитации эволюции последовательности человеческого гуанин-связывающего регуляторного белка использовался скрипт bash. Применительно к фрагменту белка из 78 нк., переведённых в а.м.о., проследим возможные изменения в семи поколениях при менее жёсткой радиации (-count 4). Выбранный для мутирования файл с участком последовательности нуклеотидов:
>M16514.1 Human guanine nucleotide-binding regulatory protein (G protein) gene, 3' end GGCCCCCGTCCCGCGGCCCCCAGCCGCCCCCAACCCTGCCCCACGGGGCCCGGCGCCATG AGTGAGCTGGAGCAACTG
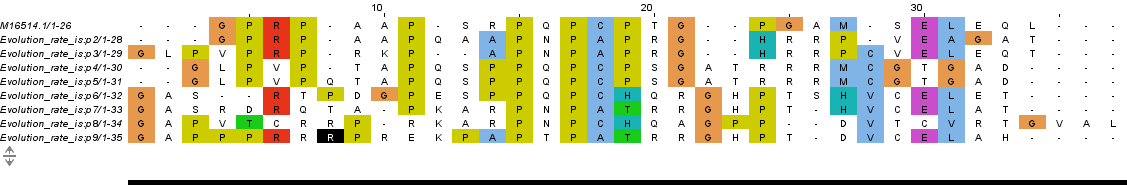
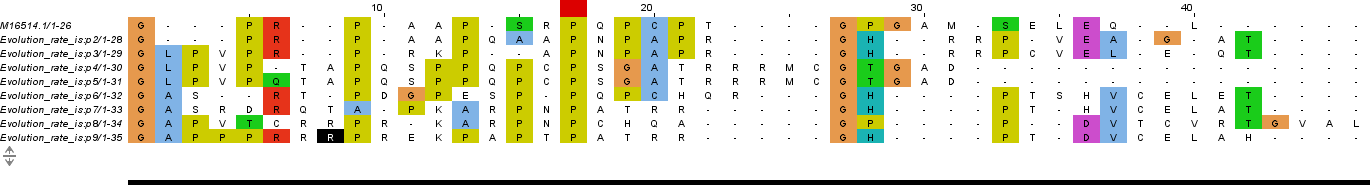
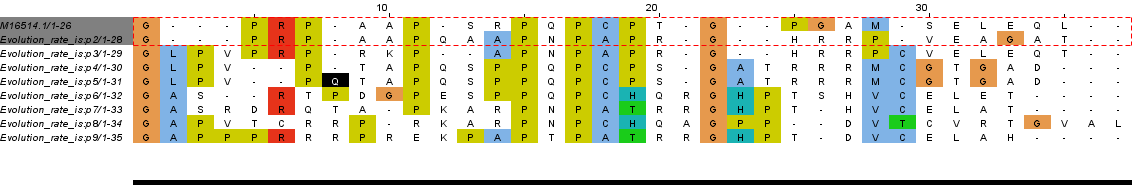
Из выравниваний видно, что, как и в случае с аминокислотной последовательностью, наблюдается большое количетво неверно выровненых столбцов. Исправлять такие выравнивания оказалось гораздо сложнее и интереснее предыдущих. Похоже, что в таких случаях программа выравнивает лучше. Интересно, что при мутировании не образоватось стоп-кодонов. Также стоит отметить, что при меньшей скорости мутирования количество мутаций сравнимо и даже превосходит предыдущий случай, что может говорить о большей чувствительности к мутациям благодаря пропорции ген. кода. Что не удивительно.
Возьму на себя смелость привести несколько контрпримеров к тезисам в презентации.
Существуют данные о наследовании метилирования или о гистоновом коде.
Умозрительно- мутировавшие гены могут захватываться ретровирусами, переноситься через плаценту и встраиваться в наследственный материал зародыша. В природе не найдено подобных механизмов, но принципиальная возможность процесса показана эксериментально. Также не стоит забывать о организмах, которые размножаются только вегетативно.
Гомологичные последовательности, как правило, представленны кодирующими и некодирующими участками. Интроны вырезаются из незрелой РНК в процессе сплайсинга, а экзоны сшиваются в зрелый продукт. Так как интроны, чаще всего, не несут ярко выраженной смысловой нагрузки, в них накапливаются мутации и содержание AT пар выше такового в экзонах.
Известны участки с повышенной скоростью мутирования. Например, кассеты антител или участки, кодирующие белки-участники иммунной системы. Подобные объекты и процессы, по- моему, обсуждаются в книгах Александра Маркова "Рождение сложности" и "Эволюция".
Конечно, вероятность потери функции белком чрезвычайно велика, но можно придумать такую модель, и, по- моему, я где- то слышал о подобном реально существующем механизме, когда рибосома скользит по иРНК, находит стартовый кодон, синтезирует часть белка, а после проскальзывает участок до следующей инициаторной или иной области. Изменение рамки считывания в промежутке может не приводить к изменению последовательности.
Как мы могли убедиться в блоке визуализации молекул, активные центры некоторых негомологичных белков имеют схожие пространственные структуры, приспособленные для выполнения схожих функций. Возникает аналогия.
© Кравченко Павел
2017