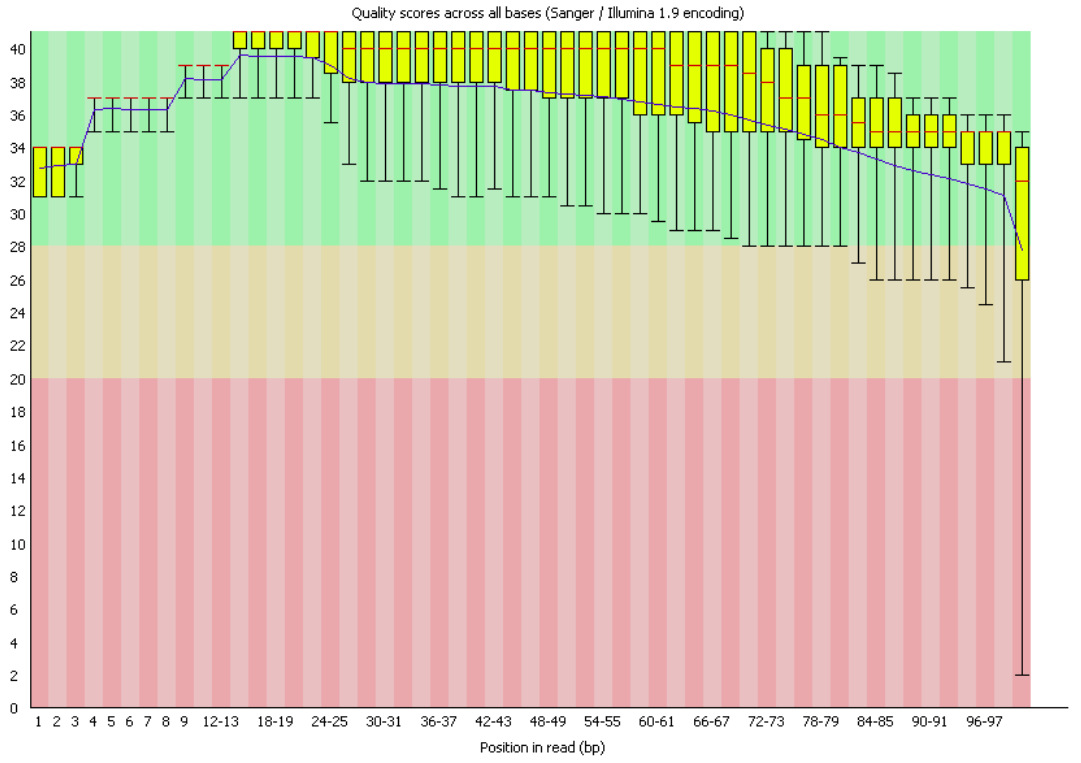
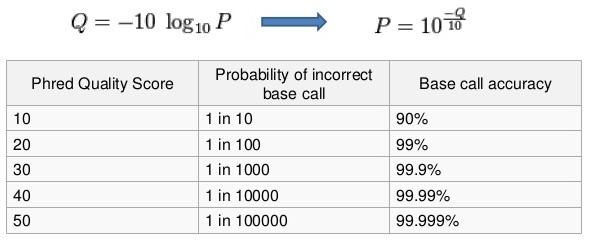
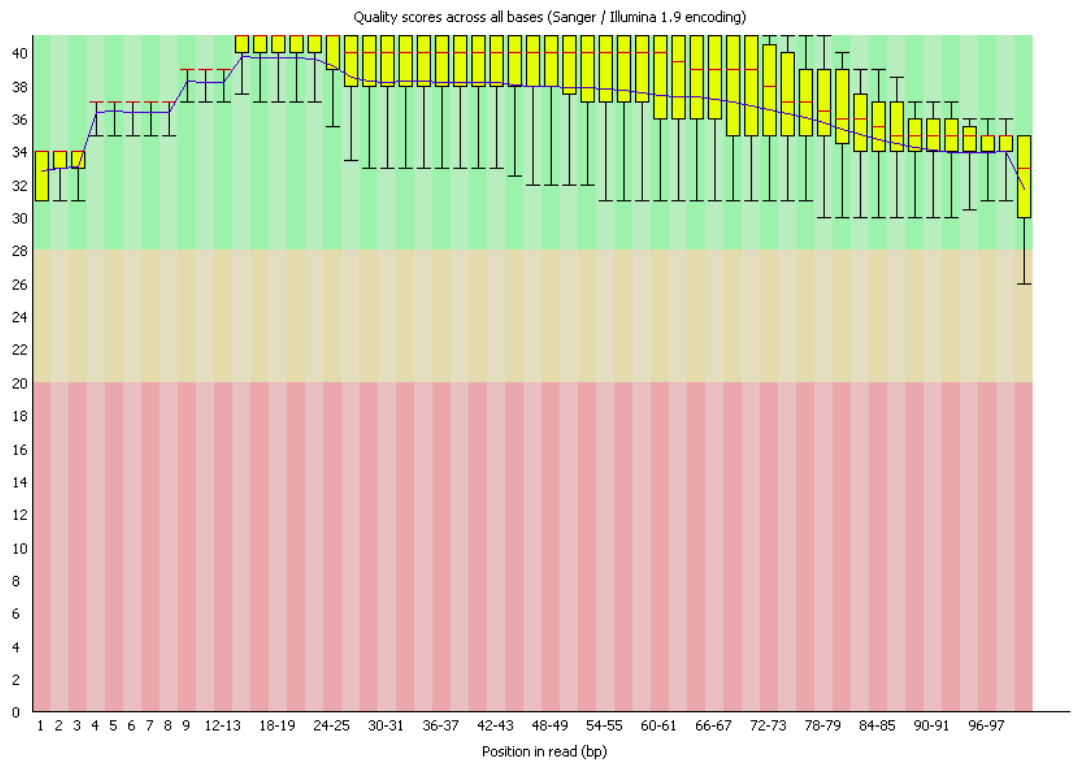
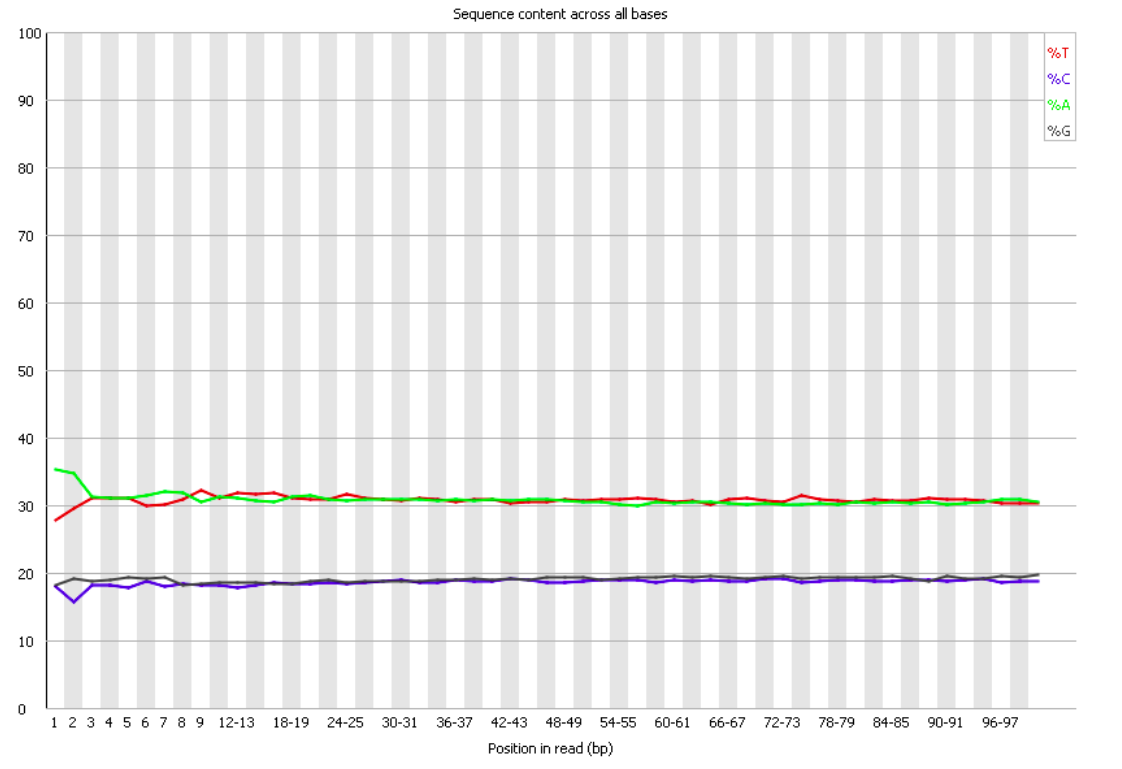
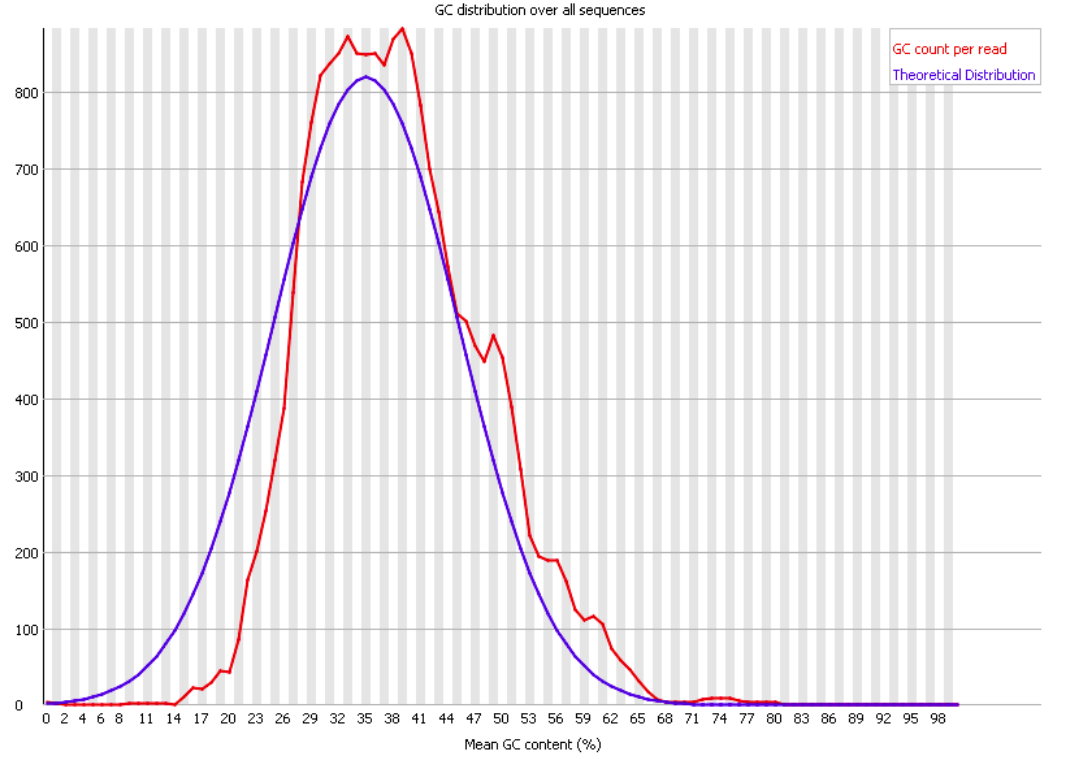
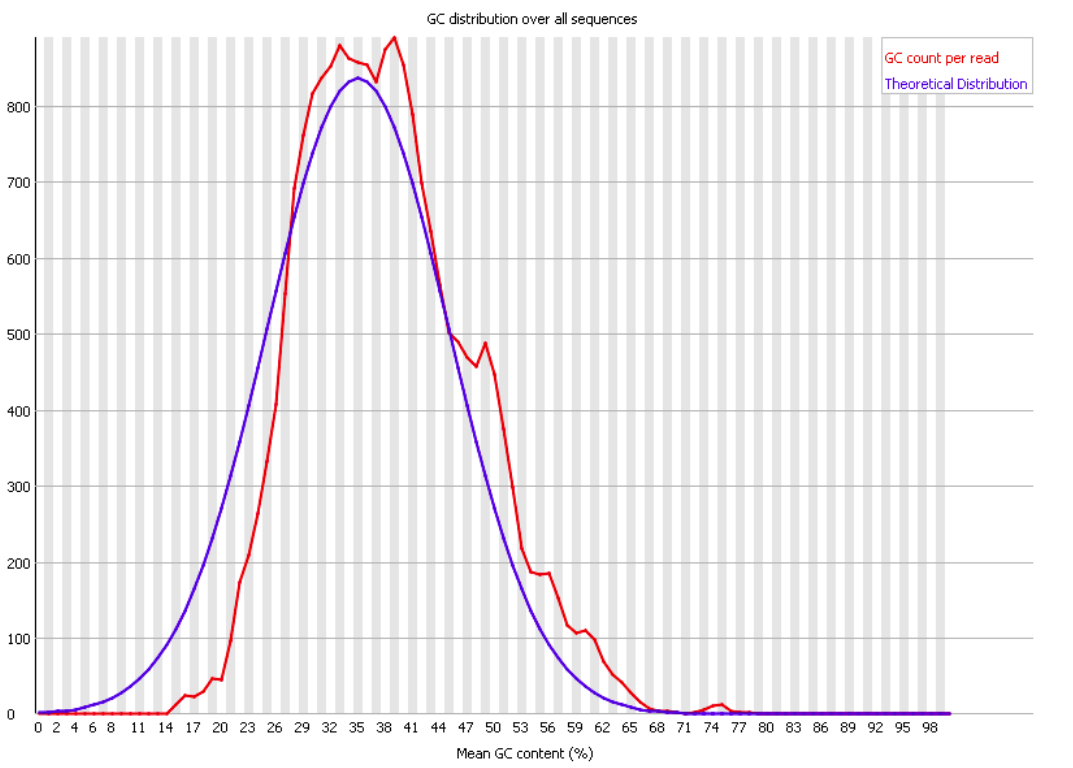
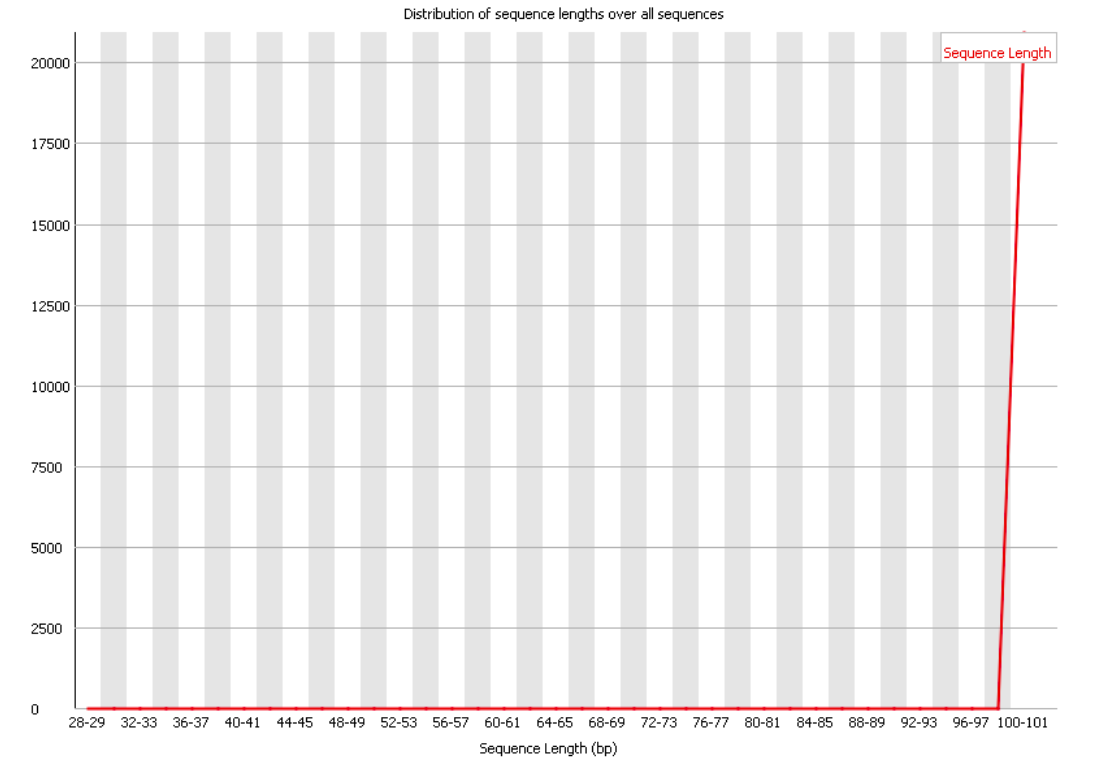
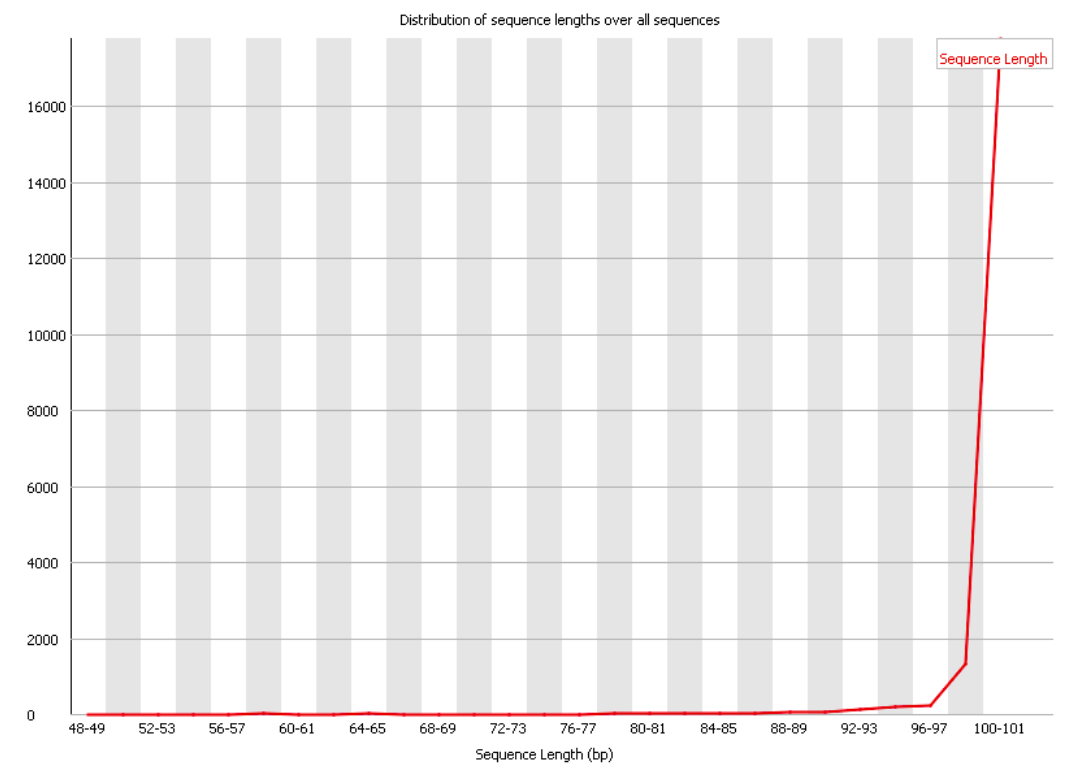
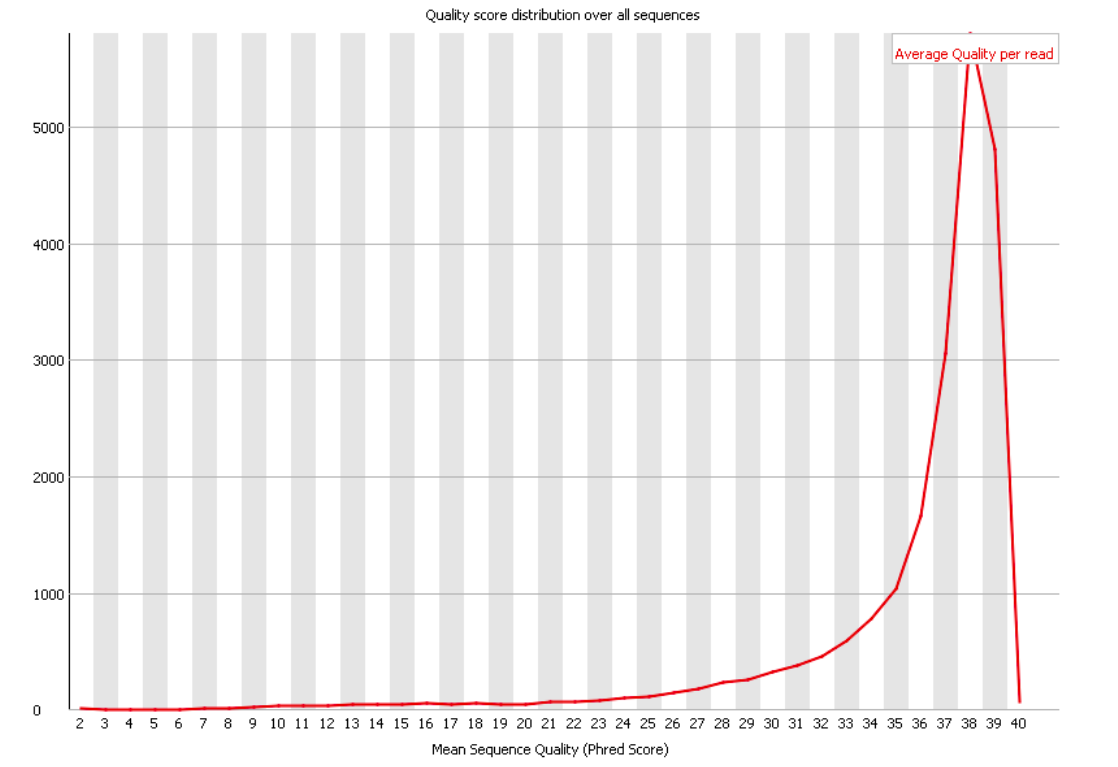
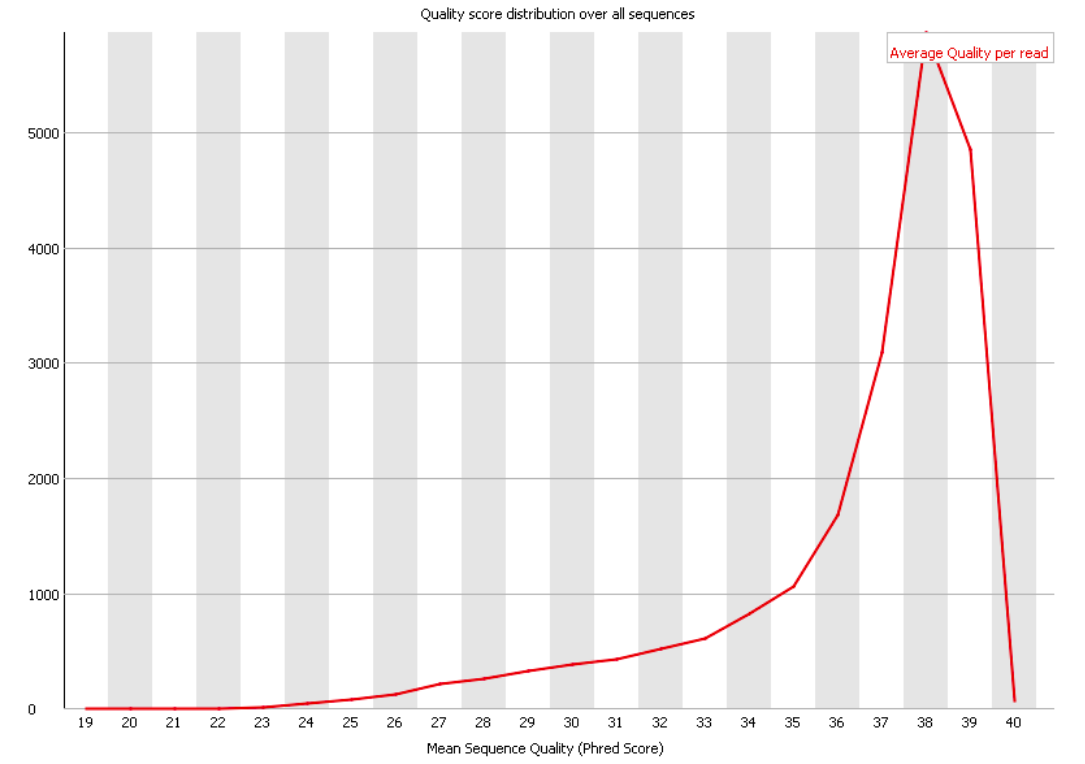
Сохранить вывод программы hisat2 в отдельный файл.
Таблица 1. Использовавшиеся команды.
| Команда |
Результат |
| export PATH=${PATH}:/home/students/y06/anastaisha_w/hisat2-2.0.5 |
Прописывает временный путь до исполняемой программы |
| hisat2-build chr3.fasta chr3 |
Индексирует референсную последовательность, записывает результат в *.ht2 файлы |
| hisat2 -x chr3 -U chr3_outfile.fastq --no-spliced-alignment --no-softclip > alignment.sam |
Строит выранивание прочтения и референса, записывает результат в .sam файл |
Программа hisat2 выдаласледующую информацию о выравниваниях.
20570 reads; of these:
20570 (100.00%) were unpaired; of these:
92 (0.45%) aligned 0 times
20467 (99.50%) aligned exactly 1 time
11 (0.05%) aligned >1 times
99.55% overall alignment rate
Было поручено 20570 ридов, из которых 92 были выровнены 0 раз, а 20467 лишь один раз; 11 были выровнены более 1 раза.
Был получен файл с выравниванием
4. Анализ выравнивания.
Для анализа выравнивания использовалась программа samtools, работающая с бинарными файлами.
Таблица 2. Использовавшиеся команды.
| Команда |
Результат |
| samtools view alignment.sam -b -o alignment.bam |
Переводит выравнивание в бинарный формат |
| samtools sort alignment.bam -T out_sort.txt -o alignment_sorted.bam |
Индексирует референсную последовательность, записывает результат в *.ht2 файлы |
| samtools index alignment_sorted.bam |
Индексирует отсортированный .bam файл |
| samtools idxstats alignment_sorted.bam > out.txt |
Записывает откартировавшиеся прочтения |
Файл out.txt содержит информацию о картировании ридов:
chr3 198022430 20489 0
* 0 0 92
Данная информация говорит о том, что на хромосому было картировано 20489 ридов, тогда как 92 ридов не было картировано. Длина хромосомы составила 198022430 нуклеотидов.
Часть III: Анализ SNP
5. Поиск SNP и инделей.
Для поиска SNP и инделей были использованы команды, представленные в таблице 3.
Таблица 3. Использовавшиеся команды.
| Команда |
Результат |
| samtools mpileup -uf chr3.fasta alignment_sorted.bam > snp.bcf |
Создаёт файл с полиморфизмами в формате .bcf |
| bcftools call -cv snp.bcf -o snp.vcf |
Создаёт файл со списком отличий между референсом и чтениями в формате .vcf |
В выдаче обнаружено более ста SNP, а также 11 инделей. Был сформирован файл, содержащий информацию о глубине прочтения каждого SNP.
Команда: samtools depth alignment_sorted.bam >depth_sorted.tsv
Cамыми интересными, на мой взгляд, оказались три, приведённые в таблице 4.
Первые два были выбраны из-за высокой глубины покрытия и одинаковым качеством прочтений, а второе из SNP с худшими параметрами наугад.
Таблица 4. Найденные SNP
| Кордината |
Tип полиморфизма: замена, вставка или делеция |
В референсной последовательности |
Что найдено в чтениях |
Глубина покрытия |
Качество прочтений |
| 41937051 |
Замена |
T |
C |
256 |
225.009 |
| 41925423 |
Замена |
T |
C |
110 |
225.009 |
| 41607701 |
Замена |
C |
G |
36 |
137.008 |
Визуализация SNPs...
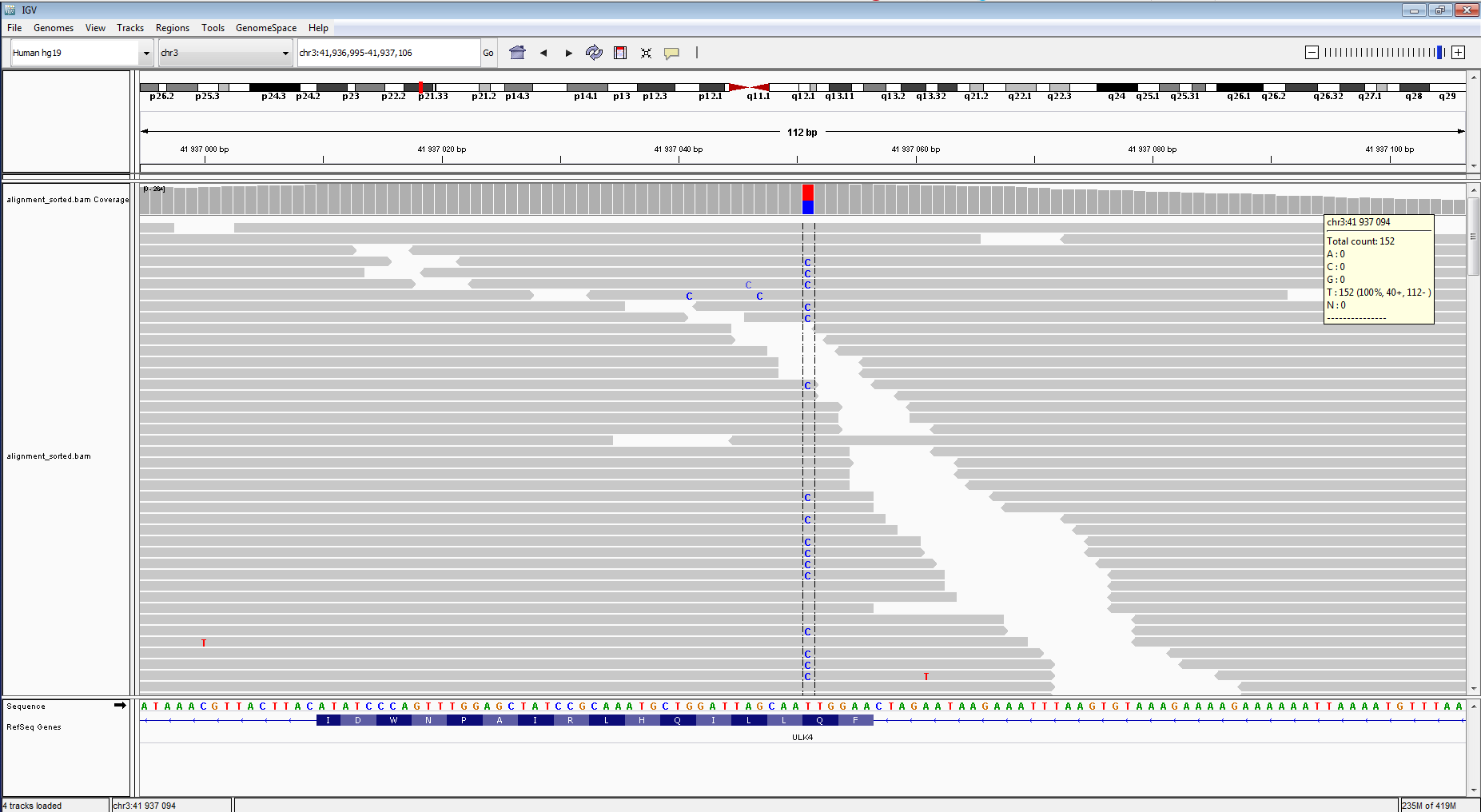
Рисунок 15. SNP в визуализаторе IGV выглядит так
|
Рисунок 15. SNP 41937051

|
|
|
|
|
|
|
|
|
|
|
|
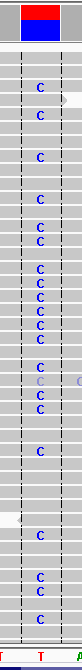
Рисунок 16. SNP 41925423
|
|
|
|
|
|
|
|
|
|
|
/ |
|
|
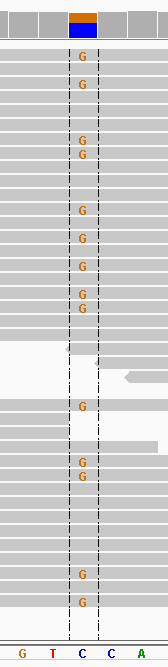
Рисунок 17. SNP 41607701
|
6. Аннотация SNP.
С помощью программы annovar были проаннотированы полученные snp. Использовались базы данных: refgene, dbsnp, 1000 genomes, GWAS, Clinvar.
Для подготовки входных файлов из .vsf файла были удалены все индели, и для очищенного файла использовался скрипт convert2annovar.pl
Команда: perl /nfs/srv/databases/annovar/convert2annovar.pl -format vcf4 /nfs/srv/databases/ngs/pavel-kravchenko/snp.vcf > /nfs/srv/databases/ngs/pavel-kravchenko/snp.avinput
Результат:
Finished reading 250 lines from VCF file
A total of 218 locus in VCF file passed QC threshold, representing 218 SNPs
(121 transitions and 97 transversions) and 1 indels/substitutions
Finished writing 217 SNP genotypes (121 transitions and 96 transversions)
and 1 indels/substitutions for 1 sample
В файл было записано 217 SNP. Для аннотации SNP с помощью баз данных была использована программа annotate_variation.pl
Аннотация по dbSNP
Был проведён поиск rc SNP по базе данных dbSNP.
Команда: /nfs/srv/databases/annovar/annotate_variation.pl -filter -out snp.rs -build hg19 -dbtype snp138 snp.avinput /nfs/srv/databases/annovar/humandb/
Были получены следующие файлы, содержащие информацию о работе и результат работы программы.
snp.rs.hg19_snp_dropped
snp.rs.hg19_snp_filtered
snp.rs.hg19_snp138_dropped
snp.rs.hg19_snp138_filtered
snp.rs.invalid_input
snp.rs.log
В файле snp.rs.hg19_snp138_dropped были записаны аннотированные SNP. 178 SNP имеют rs.
snp138 rs9290439 chr3 171768074 171768074 A G hom 11.3429 1
snp138 rs73880103 chr3 171824240 171824240 G A hom 11.3429 1
snp138 rs4894808 chr3 171833266 171833266 G C hom 6.20226 1
snp138 rs4894813 chr3 171861100 171861100 A G hom 5.46383 1
snp138 rs9816937 chr3 171863825 171863825 G A hom 42.7648 2
snp138 rs9840449 chr3 171863859 171863859 T C hom 5.46383 2
snp138 rs73880115 chr3 171881279 171881279 T C het 3.54557 1
snp138 rs13089740 chr3 171886438 171886438 T G hom 9.52546 1
snp138 rs34716963 chr3 171887000 171887000 G C het 4.13015 2
snp138 rs35796867 chr3 171895648 171895648 T C hom 6.20226 1
snp138 rs9825851 chr3 171900737 171900737 G A hom 11.3429 1
snp138 rs6445042 chr3 171910642 171910642 A G hom 11.3429 1
snp138 rs61237548 chr3 171911118 171911118 C T hom 6.20226 1
snp138 rs7646440 chr3 171916714 171916714 T C hom 9.52546 1
snp138 rs3924140 chr3 171926314 171926314 C G hom 11.3429 1
snp138 rs4535251 chr3 171926373 171926373 T C hom 7.79993 1
snp138 rs6806070 chr3 171932935 171932935 C T hom 8.64911 1
snp138 rs12629746 chr3 171933069 171933069 C A hom 44.7647 2
snp138 rs6445046 chr3 171933252 171933252 G T hom 7.79993 1
snp138 rs13077747 chr3 171949432 171949432 T C hom 11.3429 1
snp138 rs7653242 chr3 171951720 171951720 G A hom 6.20226 1
snp138 rs1039027 chr3 171965109 171965109 A G hom 184.999 24
snp138 rs7625921 chr3 171965629 171965629 A G hom 221.999 51
snp138 rs7652177 chr3 171969077 171969077 C G hom 221.999 66
snp138 rs35366330 chr3 171969228 171969228 T C hom 221.999 30
snp138 rs6796793 chr3 171984761 171984761 C T hom 11.3429 1
snp138 rs16845242 chr3 171985435 171985435 C T hom 11.3429 1
snp138 rs6445058 chr3 172016000 172016000 G T hom 7.79993 1
snp138 rs73880143 chr3 172016753 172016753 G A hom 6.98265 2
snp138 rs6795618 chr3 172045772 172045772 A T hom 11.3429 1
snp138 rs2270568 chr3 172046861 172046861 T C het 225.009 95
snp138 rs6799133 chr3 172046933 172046933 A G het 225.009 87
snp138 rs60375808 chr3 172055273 172055273 T C het 118.008 25
snp138 rs502170 chr3 172064828 172064828 A C het 149.008 35
snp138 rs6786014 chr3 172082479 172082479 C G hom 9.52546 1
snp138 rs73880156 chr3 172090650 172090650 A T hom 9.52546 1
snp138 rs6798409 chr3 172095880 172095880 C A het 54.0072 8
snp138 rs596590 chr3 172114474 172114474 G C hom 43.7647 2
snp138 rs596523 chr3 172114520 172114520 C T hom 11.3429 1
snp138 rs6794474 chr3 172115465 172115465 T A het 161.009 48
snp138 rs494572 chr3 172115466 172115466 T A het 164.009 47
snp138 rs495702 chr3 172115640 172115640 A T het 101.008 11
snp138 rs73880159 chr3 172116485 172116485 C T hom 6.20226 1
snp138 rs2293304 chr3 41291081 41291081 G A hom 221.999 25
snp138 rs13434088 chr3 41320126 41320126 T C hom 8.64911 1
snp138 rs72863428 chr3 41336682 41336682 T C hom 11.3429 1
snp138 rs3912578 chr3 41349781 41349781 A T hom 9.52546 1
snp138 rs1487568 chr3 41349977 41349977 G A hom 9.52546 1
snp138 rs4973937 chr3 41359534 41359534 A G hom 6.98265 1
snp138 rs6770252 chr3 41364241 41364241 G A hom 10.4247 1
snp138 rs1256687 chr3 41371717 41371717 T C hom 38.7651 2
snp138 rs9820239 chr3 41372090 41372090 C T hom 10.4247 1
snp138 rs1532015 chr3 41375030 41375030 C G hom 9.52546 1
snp138 rs9857557 chr3 41375199 41375199 G T hom 6.98265 1
snp138 rs9857720 chr3 41375275 41375275 G C hom 7.79993 1
snp138 rs749931 chr3 41378582 41378582 C A hom 11.3429 1
snp138 rs749932 chr3 41378621 41378621 T C hom 11.3429 1
snp138 rs73831321 chr3 41379045 41379045 C T hom 6.20226 1
snp138 rs73831322 chr3 41379048 41379048 A T hom 6.20226 1
snp138 rs56259019 chr3 41398724 41398724 T C hom 11.3429 1
snp138 rs56908202 chr3 41400196 41400196 C T hom 39.765 2
snp138 rs59525888 chr3 41400697 41400697 T C hom 11.3429 1
snp138 rs60482228 chr3 41400713 41400713 C T hom 38.7651 2
snp138 rs56157678 chr3 41400717 41400717 T C hom 40.7649 2
snp138 rs75500851 chr3 41402366 41402366 T C hom 8.64911 1
snp138 rs61207466 chr3 41402415 41402415 G A hom 38.7651 2
snp138 rs73828006 chr3 41404892 41404892 G A hom 9.52546 1
snp138 rs7622755 chr3 41408203 41408203 C T het 3.01618 1
snp138 rs9311273 chr3 41410711 41410711 C A hom 11.3429 1
snp138 rs7640562 chr3 41412741 41412741 G C hom 10.4247 1
snp138 rs9824480 chr3 41443267 41443267 C T het 70.0075 5
snp138 rs34196069 chr3 41452686 41452686 A C hom 11.3429 1
snp138 rs78804073 chr3 41470865 41470865 T C hom 11.3429 1
snp138 rs73828024 chr3 41477072 41477072 T C hom 8.64911 1
snp138 rs12152500 chr3 41478258 41478258 C A hom 11.3429 1
snp138 rs2270605 chr3 41496752 41496752 A C hom 64.9723 3
snp138 rs9841301 chr3 41497165 41497165 G A het 104.008 16
snp138 rs73828041 chr3 41498861 41498861 A C hom 11.3429 1
snp138 rs59838973 chr3 41502917 41502917 T C hom 7.79993 1
snp138 rs73828047 chr3 41504679 41504679 T C het 88.0076 8
snp138 rs9845791 chr3 41527905 41527905 C T hom 7.79993 1
snp138 rs9846397 chr3 41528100 41528100 G T hom 9.52546 1
snp138 rs1390340 chr3 41528106 41528106 C T hom 11.3429 1
snp138 rs12639280 chr3 41540052 41540052 C T hom 8.64911 1
snp138 rs73828090 chr3 41542282 41542282 C T hom 10.4247 1
snp138 rs1267088 chr3 41549724 41549724 T C het 3.54366 2
snp138 rs1691977 chr3 41554290 41554290 G A het 4.77219 1
snp138 rs1874354 chr3 41557727 41557727 A T hom 11.3429 1
snp138 rs6765331 chr3 41570382 41570382 C A hom 8.64911 1
snp138 rs1691966 chr3 41589964 41589964 G C hom 7.79993 1
snp138 rs1795359 chr3 41590054 41590054 C T hom 7.79993 1
snp138 rs1795360 chr3 41590068 41590068 C T hom 11.3429 1
snp138 rs2088192 chr3 41606166 41606166 C T hom 7.79993 1
snp138 rs2700448 chr3 41606170 41606170 A G hom 9.52546 1
snp138 rs2700447 chr3 41606251 41606251 T G hom 38.7651 2
snp138 rs60217205 chr3 41606466 41606466 C T hom 11.3429 1
snp138 rs6786299 chr3 41607388 41607388 T C het 120.008 27
snp138 rs73830238 chr3 41607450 41607450 C T het 222.009 52
snp138 rs1565957 chr3 41607701 41607701 C G het 137.008 36
snp138 rs17058895 chr3 41615845 41615845 T G het 4.13164 1
snp138 rs62256950 chr3 41615913 41615913 C T hom 6.20226 2
snp138 rs79506506 chr3 41635519 41635519 G C hom 11.3429 1
snp138 rs4973976 chr3 41638984 41638984 G A hom 11.3429 1
snp138 rs72862121 chr3 41658278 41658278 C T hom 9.52546 1
snp138 rs72862123 chr3 41658297 41658297 G A hom 34.7659 2
snp138 rs6775563 chr3 41669123 41669123 T C hom 11.3429 1
snp138 rs10510724 chr3 41681725 41681725 C T het 5.46063 2
snp138 rs57189600 chr3 41683734 41683734 G A hom 10.4247 1
snp138 rs57493405 chr3 41737481 41737481 T C hom 7.79993 1
snp138 rs112931149 chr3 41737598 41737598 A G hom 11.3429 2
snp138 rs28661858 chr3 41738596 41738596 C T hom 8.64911 1
snp138 rs9812782 chr3 41741505 41741505 C T hom 11.3429 1
snp138 rs9850380 chr3 41741514 41741514 A T hom 10.4247 1
snp138 rs55712089 chr3 41748938 41748938 A C hom 11.3429 1
snp138 rs6599168 chr3 41750323 41750323 T C hom 11.3429 1
snp138 rs61744388 chr3 41756965 41756965 C T hom 222 38
snp138 rs61744385 chr3 41756986 41756986 A T hom 222.085 30
snp138 rs17060938 chr3 41759191 41759191 T C hom 220.999 34
snp138 rs73828236 chr3 41759457 41759457 A G het 62.0073 9
snp138 rs9852303 chr3 41759525 41759525 C T hom 62.9723 3
snp138 rs60061241 chr3 41779622 41779622 A T het 4.13164 1
snp138 rs61672388 chr3 41784422 41784422 A G hom 35.7656 2
snp138 rs9867627 chr3 41795841 41795841 A C het 216.009 19
snp138 rs79548696 chr3 41803385 41803385 C T hom 9.52546 1
snp138 rs9837273 chr3 41813104 41813104 T C hom 43.7647 2
snp138 rs9817510 chr3 41813108 41813108 C T hom 39.765 2
snp138 rs35338196 chr3 41824696 41824696 - T het 4.4191 1
snp138 rs17215589 chr3 41831203 41831203 C T het 206.009 30
snp138 rs12186051 chr3 41836919 41836919 G A hom 7.79993 1
snp138 rs12186109 chr3 41836920 41836920 T C hom 10.4247 1
snp138 rs4973986 chr3 41841716 41841716 A C hom 221.999 31
snp138 rs17215883 chr3 41841873 41841873 C T het 78.0075 12
snp138 rs73073368 chr3 41843391 41843391 A G hom 7.79993 1
snp138 rs73073371 chr3 41843443 41843443 A G hom 11.3429 1
snp138 rs113683976 chr3 41850685 41850685 A C het 3.54557 1
snp138 rs17063572 chr3 41860955 41860955 A G het 171.009 25
snp138 rs17063653 chr3 41863817 41863817 A G hom 9.52546 1
snp138 rs73079316 chr3 41863859 41863859 G A hom 11.3429 1
snp138 rs73079327 chr3 41864697 41864697 A T het 6.20096 2
snp138 rs6790732 chr3 41864872 41864872 C T hom 11.3429 1
snp138 rs150594648 chr3 41869847 41869847 A T hom 6.98265 1
snp138 rs77717550 chr3 41872504 41872504 T A hom 10.4247 1
snp138 rs9816560 chr3 41872527 41872527 G T hom 7.79993 1
snp138 rs3774372 chr3 41877414 41877414 T C het 205.009 43
snp138 rs114960066 chr3 41882323 41882323 A G hom 6.20226 1
snp138 rs146707962 chr3 41882357 41882357 T C het 4.13164 1
snp138 rs11921993 chr3 41886884 41886884 A C hom 11.3429 1
snp138 rs7610291 chr3 41888671 41888671 G C het 4.77219 1
snp138 rs2683698 chr3 41900656 41900656 A G het 62.0073 10
snp138 rs2683699 chr3 41900774 41900774 T C het 181.009 16
snp138 rs2625667 chr3 41900951 41900951 C A hom 221.999 24
snp138 rs73830534 chr3 41901030 41901030 C A het 225.009 32
snp138 rs1717028 chr3 41902964 41902964 C T hom 11.3429 1
snp138 rs1716674 chr3 41902972 41902972 G C hom 11.3429 1
snp138 rs9832548 chr3 41903491 41903491 A T hom 11.3429 1
snp138 rs1717029 chr3 41903601 41903601 G T hom 10.4247 1
snp138 rs1717030 chr3 41904576 41904576 A T hom 7.79993 1
snp138 rs1717032 chr3 41909219 41909219 C T het 4.13164 1
snp138 rs1716670 chr3 41910571 41910571 T C hom 10.4247 1
snp138 rs1716994 chr3 41923872 41923872 T C het 106.008 12
snp138 rs1716687 chr3 41925423 41925423 T C het 225.009 110
snp138 rs73087378 chr3 41936827 41936827 G A het 147.008 91
snp138 rs73830560 chr3 41937051 41937051 T C het 225.009 256
snp138 rs1717005 chr3 41938500 41938500 G C het 168.009 21
snp138 rs73069209 chr3 41939781 41939781 G A het 167.009 37
snp138 rs1625226 chr3 41939993 41939993 T A het 225.009 67
snp138 rs59146151 chr3 41949301 41949301 T C het 134.008 16
snp138 rs1716991 chr3 41952781 41952781 C T het 225.009 95
snp138 rs35263917 chr3 41952852 41952852 T C het 225.009 76
snp138 rs1716992 chr3 41973062 41973062 T C het 5.46063 2
snp138 rs73071203 chr3 41977464 41977464 A C het 225.009 51
snp138 rs73071208 chr3 41978738 41978738 A T het 194.009 29
snp138 rs1716691 chr3 41983615 41983615 G C het 5.46063 2
snp138 rs7651623 chr3 41996275 41996275 A G het 225.009 47
snp138 rs17284472 chr3 41996304 41996304 G A het 217.009 36
snp138 rs1108842 chr3 52720080 52720080 A C hom 221.999 74
snp138 rs72960212 chr3 52720489 52720489 T A hom 11.3429 1
snp138 rs3774349 chr3 52722335 52722335 A C het 100.062 8
Аннотация по RefGene
Была выполнена аннотация по базе данных RefGene. По аннотации можно судить, в какой из функционально или топологически нагруженных участков попал найденный SNP.
Команда: perl /nfs/srv/databases/annovar/annotate_variation.pl -out snp.refgene -build hg19 snp.avinput /nfs/srv/databases/annovar/humandb/
Результаты работы программы можно найти в директории, а также посмотреть распределение SNP по группам в таблице 5.
snp.refgene.exonic_variant_function
snp.refgene.log
snp.refgene.variant_function
intronic ULK4 chr3 41291081 41291081 G A hom 221.999 25
intronic ULK4 chr3 41309334 41309334 C A het 4.12848 3
intronic ULK4 chr3 41310211 41310211 G A hom 5.46383 1
intronic ULK4 chr3 41310663 41310663 A C het 4.12954 2
intronic ULK4 chr3 41320126 41320126 T C hom 8.64911 1
intronic ULK4 chr3 41324367 41324367 C T hom 9.52546 1
intronic ULK4 chr3 41336682 41336682 T C hom 11.3429 1
intronic ULK4 chr3 41349781 41349781 A T hom 9.52546 1
intronic ULK4 chr3 41349977 41349977 G A hom 9.52546 1
intronic ULK4 chr3 41359534 41359534 A G hom 6.98265 1
intronic ULK4 chr3 41360231 41360231 G T hom 9.52546 1
intronic ULK4 chr3 41362118 41362118 C A hom 5.46383 1
intronic ULK4 chr3 41364241 41364241 G A hom 10.4247 1
intronic ULK4 chr3 41371717 41371717 T C hom 38.7651 2
intronic ULK4 chr3 41372090 41372090 C T hom 10.4247 1
intronic ULK4 chr3 41375030 41375030 C G hom 9.52546 1
intronic ULK4 chr3 41375199 41375199 G T hom 6.98265 1
intronic ULK4 chr3 41375275 41375275 G C hom 7.79993 1
intronic ULK4 chr3 41378582 41378582 C A hom 11.3429 1
intronic ULK4 chr3 41378621 41378621 T C hom 11.3429 1
intronic ULK4 chr3 41379045 41379045 C T hom 6.20226 1
intronic ULK4 chr3 41379048 41379048 A T hom 6.20226 1
intronic ULK4 chr3 41398724 41398724 T C hom 11.3429 1
intronic ULK4 chr3 41400196 41400196 C T hom 39.765 2
intronic ULK4 chr3 41400697 41400697 T C hom 11.3429 1
intronic ULK4 chr3 41400713 41400713 C T hom 38.7651 2
intronic ULK4 chr3 41400717 41400717 T C hom 40.7649 2
intronic ULK4 chr3 41402366 41402366 T C hom 8.64911 1
intronic ULK4 chr3 41402415 41402415 G A hom 38.7651 2
intronic ULK4 chr3 41404892 41404892 G A hom 9.52546 1
intronic ULK4 chr3 41408203 41408203 C T het 3.01618 1
intronic ULK4 chr3 41408210 41408210 T A hom 5.46383 1
intronic ULK4 chr3 41410711 41410711 C A hom 11.3429 1
intronic ULK4 chr3 41412741 41412741 G C hom 10.4247 1
intronic ULK4 chr3 41423613 41423613 C A hom 10.4247 1
intronic ULK4 chr3 41424542 41424542 A G het 3.54557 1
intronic ULK4 chr3 41443267 41443267 C T het 70.0075 5
intronic ULK4 chr3 41452686 41452686 A C hom 11.3429 1
intronic ULK4 chr3 41459943 41459943 G T hom 6.20226 1
intronic ULK4 chr3 41470865 41470865 T C hom 11.3429 1
intronic ULK4 chr3 41477072 41477072 T C hom 8.64911 1
intronic ULK4 chr3 41478258 41478258 C A hom 11.3429 1
intronic ULK4 chr3 41496752 41496752 A C hom 64.9723 3
intronic ULK4 chr3 41497165 41497165 G A het 104.008 16
intronic ULK4 chr3 41498861 41498861 A C hom 11.3429 1
intronic ULK4 chr3 41499103 41499103 C T hom 27.7716 2
intronic ULK4 chr3 41502917 41502917 T C hom 7.79993 1
intronic ULK4 chr3 41504513 41504513 G T het 9.52088 11
exonic ULK4 chr3 41504679 41504679 T C het 88.0076 8
intronic ULK4 chr3 41504887 41504887 G T het 4.76921 2
intronic ULK4 chr3 41512465 41512465 A C hom 7.79993 1
intronic ULK4 chr3 41527905 41527905 C T hom 7.79993 1
intronic ULK4 chr3 41528100 41528100 G T hom 9.52546 1
intronic ULK4 chr3 41528106 41528106 C T hom 11.3429 1
intronic ULK4 chr3 41540052 41540052 C T hom 8.64911 1
intronic ULK4 chr3 41542282 41542282 C T hom 10.4247 1
intronic ULK4 chr3 41549724 41549724 T C het 3.54366 2
intronic ULK4 chr3 41554290 41554290 G A het 4.77219 1
intronic ULK4 chr3 41557727 41557727 A T hom 11.3429 1
intronic ULK4 chr3 41565131 41565131 G T hom 11.3429 1
intronic ULK4 chr3 41570382 41570382 C A hom 8.64911 1
intronic ULK4 chr3 41581286 41581286 C A hom 9.52546 1
intronic ULK4 chr3 41589964 41589964 G C hom 7.79993 1
intronic ULK4 chr3 41590054 41590054 C T hom 7.79993 1
intronic ULK4 chr3 41590068 41590068 C T hom 11.3429 1
intronic ULK4 chr3 41606166 41606166 C T hom 7.79993 1
intronic ULK4 chr3 41606170 41606170 A G hom 9.52546 1
intronic ULK4 chr3 41606251 41606251 T G hom 38.7651 2
intronic ULK4 chr3 41606466 41606466 C T hom 11.3429 1
intronic ULK4 chr3 41607388 41607388 T C het 120.008 27
intronic ULK4 chr3 41607450 41607450 C T het 222.009 52
intronic ULK4 chr3 41607701 41607701 C G het 137.008 36
intronic ULK4 chr3 41615845 41615845 T G het 4.13164 1
intronic ULK4 chr3 41615913 41615913 C T hom 6.20226 2
intronic ULK4 chr3 41617657 41617657 C A hom 8.64911 1
intronic ULK4 chr3 41635519 41635519 G C hom 11.3429 1
intronic ULK4 chr3 41638984 41638984 G A hom 11.3429 1
intronic ULK4 chr3 41658278 41658278 C T hom 9.52546 1
intronic ULK4 chr3 41658297 41658297 G A hom 34.7659 2
intronic ULK4 chr3 41669123 41669123 T C hom 11.3429 1
intronic ULK4 chr3 41681725 41681725 C T het 5.46063 2
intronic ULK4 chr3 41683734 41683734 G A hom 10.4247 1
intronic ULK4 chr3 41720865 41720865 G T hom 7.79993 1
intronic ULK4 chr3 41737481 41737481 T C hom 7.79993 1
intronic ULK4 chr3 41737598 41737598 A G hom 11.3429 2
intronic ULK4 chr3 41738596 41738596 C T hom 8.64911 1
intronic ULK4 chr3 41741505 41741505 C T hom 11.3429 1
intronic ULK4 chr3 41741514 41741514 A T hom 10.4247 1
intronic ULK4 chr3 41742413 41742413 G T hom 11.3429 1
intronic ULK4 chr3 41748938 41748938 A C hom 11.3429 1
intronic ULK4 chr3 41750323 41750323 T C hom 11.3429 1
exonic ULK4 chr3 41756965 41756965 C T hom 222 38
exonic ULK4 chr3 41756986 41756986 A T hom 222.085 30
intronic ULK4 chr3 41759191 41759191 T C hom 220.999 34
intronic ULK4 chr3 41759457 41759457 A G het 62.0073 9
intronic ULK4 chr3 41759525 41759525 C T hom 62.9723 3
intronic ULK4 chr3 41768977 41768977 G A hom 8.64911 1
intronic ULK4 chr3 41779622 41779622 A T het 4.13164 1
intronic ULK4 chr3 41784422 41784422 A G hom 35.7656 2
intronic ULK4 chr3 41795841 41795841 A C het 216.009 19
intronic ULK4 chr3 41803385 41803385 C T hom 9.52546 1
intronic ULK4 chr3 41813104 41813104 T C hom 43.7647 2
intronic ULK4 chr3 41813108 41813108 C T hom 39.765 2
intronic ULK4 chr3 41824696 41824696 - T het 4.4191 1
intronic ULK4 chr3 41831088 41831088 G T het 7.79563 12
exonic ULK4 chr3 41831203 41831203 C T het 206.009 30
intronic ULK4 chr3 41836919 41836919 G A hom 7.79993 1
intronic ULK4 chr3 41836920 41836920 T C hom 10.4247 1
exonic ULK4 chr3 41841716 41841716 A C hom 221.999 31
intronic ULK4 chr3 41841873 41841873 C T het 78.0075 12
intronic ULK4 chr3 41843391 41843391 A G hom 7.79993 1
intronic ULK4 chr3 41843443 41843443 A G hom 11.3429 1
intronic ULK4 chr3 41850685 41850685 A C het 3.54557 1
intronic ULK4 chr3 41858544 41858544 G T hom 6.20226 1
exonic ULK4 chr3 41860955 41860955 A G het 171.009 25
intronic ULK4 chr3 41862536 41862536 G T hom 9.52546 1
intronic ULK4 chr3 41863817 41863817 A G hom 9.52546 1
intronic ULK4 chr3 41863859 41863859 G A hom 11.3429 1
intronic ULK4 chr3 41864697 41864697 A T het 6.20096 2
intronic ULK4 chr3 41864872 41864872 C T hom 11.3429 1
intronic ULK4 chr3 41869847 41869847 A T hom 6.98265 1
intronic ULK4 chr3 41872504 41872504 T A hom 10.4247 1
intronic ULK4 chr3 41872527 41872527 G T hom 7.79993 1
exonic ULK4 chr3 41877414 41877414 T C het 205.009 43
intronic ULK4 chr3 41882323 41882323 A G hom 6.20226 1
intronic ULK4 chr3 41882340 41882340 T A hom 6.20226 1
intronic ULK4 chr3 41882357 41882357 T C het 4.13164 1
intronic ULK4 chr3 41886884 41886884 A C hom 11.3429 1
intronic ULK4 chr3 41888671 41888671 G C het 4.77219 1
intronic ULK4 chr3 41900656 41900656 A G het 62.0073 10
intronic ULK4 chr3 41900774 41900774 T C het 181.009 16
intronic ULK4 chr3 41900951 41900951 C A hom 221.999 24
intronic ULK4 chr3 41901030 41901030 C A het 225.009 32
intronic ULK4 chr3 41902964 41902964 C T hom 11.3429 1
intronic ULK4 chr3 41902972 41902972 G C hom 11.3429 1
intronic ULK4 chr3 41903491 41903491 A T hom 11.3429 1
intronic ULK4 chr3 41903601 41903601 G T hom 10.4247 1
intronic ULK4 chr3 41904488 41904488 T C hom 6.98265 1
intronic ULK4 chr3 41904576 41904576 A T hom 7.79993 1
intronic ULK4 chr3 41909219 41909219 C T het 4.13164 1
intronic ULK4 chr3 41910571 41910571 T C hom 10.4247 1
intronic ULK4 chr3 41923491 41923491 C A hom 10.4247 1
intronic ULK4 chr3 41923872 41923872 T C het 106.008 12
exonic ULK4 chr3 41925423 41925423 T C het 225.009 110
intronic ULK4 chr3 41936577 41936577 G C hom 11.3429 1
intronic ULK4 chr3 41936827 41936827 G A het 147.008 91
exonic ULK4 chr3 41937051 41937051 T C het 225.009 256
intronic ULK4 chr3 41938500 41938500 G C het 168.009 21
intronic ULK4 chr3 41938751 41938751 G T hom 10.4247 1
intronic ULK4 chr3 41939781 41939781 G A het 167.009 37
intronic ULK4 chr3 41939993 41939993 T A het 225.009 67
intronic ULK4 chr3 41949301 41949301 T C het 134.008 16
intronic ULK4 chr3 41952781 41952781 C T het 225.009 95
exonic ULK4 chr3 41952852 41952852 T C het 225.009 76
intronic ULK4 chr3 41959698 41959698 C A het 4.12848 3
intronic ULK4 chr3 41973062 41973062 T C het 5.46063 2
intronic ULK4 chr3 41975058 41975058 C A hom 6.20226 1
intronic ULK4 chr3 41977464 41977464 A C het 225.009 51
intronic ULK4 chr3 41978738 41978738 A T het 194.009 29
intronic ULK4 chr3 41983615 41983615 G C het 5.46063 2
UTR5 ULK4(NM_017886:c.-24T>C) chr3 41996275 41996275 A G het 225.009 47
intronic ULK4 chr3 41996304 41996304 G A het 217.009 36
UTR5 GNL3(NM_014366:c.-29A>C,NM_206825:c.-728A>C) chr3 52720080 52720080 A C hom 221.999 74
intronic GNL3 chr3 52720489 52720489 T A hom 11.3429 1
intronic GNL3 chr3 52722335 52722335 A C het 100.062 8
intronic CADM2 chr3 85740718 85740718 C A hom 6.98265 1
intronic CADM2 chr3 85740765 85740765 C T hom 11.3429 1
intronic FNDC3B chr3 171768074 171768074 A G hom 11.3429 1
intronic FNDC3B chr3 171779010 171779010 G T hom 6.20226 1
intronic FNDC3B chr3 171824240 171824240 G A hom 11.3429 1
intronic FNDC3B chr3 171833266 171833266 G C hom 6.20226 1
intronic FNDC3B chr3 171842762 171842762 G T hom 11.3429 1
intronic FNDC3B chr3 171846907 171846907 C T het 4.76948 2
intronic FNDC3B chr3 171861100 171861100 A G hom 5.46383 1
intronic FNDC3B chr3 171863825 171863825 G A hom 42.7648 2
intronic FNDC3B chr3 171863859 171863859 T C hom 5.46383 2
intronic FNDC3B chr3 171881279 171881279 T C het 3.54557 1
intronic FNDC3B chr3 171881411 171881411 G T hom 7.79993 1
intronic FNDC3B chr3 171886438 171886438 T G hom 9.52546 1
intronic FNDC3B chr3 171887000 171887000 G C het 4.13015 2
intronic FNDC3B chr3 171895648 171895648 T C hom 6.20226 1
intronic FNDC3B chr3 171900737 171900737 G A hom 11.3429 1
intronic FNDC3B chr3 171910642 171910642 A G hom 11.3429 1
intronic FNDC3B chr3 171911118 171911118 C T hom 6.20226 1
intronic FNDC3B chr3 171916714 171916714 T C hom 9.52546 1
intronic FNDC3B chr3 171926314 171926314 C G hom 11.3429 1
intronic FNDC3B chr3 171926373 171926373 T C hom 7.79993 1
intronic FNDC3B chr3 171932935 171932935 C T hom 8.64911 1
intronic FNDC3B chr3 171933069 171933069 C A hom 44.7647 2
intronic FNDC3B chr3 171933252 171933252 G T hom 7.79993 1
intronic FNDC3B chr3 171949432 171949432 T C hom 11.3429 1
intronic FNDC3B chr3 171951720 171951720 G A hom 6.20226 1
intronic FNDC3B chr3 171965109 171965109 A G hom 184.999 24
intronic FNDC3B chr3 171965629 171965629 A G hom 221.999 51
exonic FNDC3B chr3 171969077 171969077 C G hom 221.999 66
exonic FNDC3B chr3 171969228 171969228 T C hom 221.999 30
intronic FNDC3B chr3 171981756 171981756 G C hom 11.3429 1
intronic FNDC3B chr3 171984761 171984761 C T hom 11.3429 1
intronic FNDC3B chr3 171985435 171985435 C T hom 11.3429 1
intronic FNDC3B chr3 171989504 171989504 G T het 3.01497 2
intronic FNDC3B chr3 172012129 172012129 G T hom 11.3429 1
intronic FNDC3B chr3 172016000 172016000 G T hom 7.79993 1
intronic FNDC3B chr3 172016753 172016753 G A hom 6.98265 2
intronic FNDC3B chr3 172025409 172025409 G T het 4.12848 3
intronic FNDC3B chr3 172045772 172045772 A T hom 11.3429 1
exonic FNDC3B chr3 172046861 172046861 T C het 225.009 95
intronic FNDC3B chr3 172046933 172046933 A G het 225.009 87
intronic FNDC3B chr3 172055273 172055273 T C het 118.008 25
intronic FNDC3B chr3 172064828 172064828 A C het 149.008 35
intronic FNDC3B chr3 172082479 172082479 C G hom 9.52546 1
intronic FNDC3B chr3 172090650 172090650 A T hom 9.52546 1
intronic FNDC3B chr3 172095880 172095880 C A het 54.0072 8
intronic FNDC3B chr3 172114474 172114474 G C hom 43.7647 2
intronic FNDC3B chr3 172114520 172114520 C T hom 11.3429 1
UTR3 FNDC3B(NM_022763:c.*200T>A,NM_001135095:c.*200T>A) chr3 172115465 172115465 T A het 161.009 48
UTR3 FNDC3B(NM_022763:c.*201T>A,NM_001135095:c.*201T>A) chr3 172115466 172115466 T A het 164.009 47
UTR3 FNDC3B(NM_022763:c.*375A>T,NM_001135095:c.*375A>T) chr3 172115640 172115640 A T het 101.008 11
UTR3 FNDC3B(NM_022763:c.*1220C>T,NM_001135095:c.*1220C>T) chr3 172116485 172116485 C T hom 6.20226 1
Для анализа файла может пригодиться таблица 5.
Таблица 5. Расположение SNP по гуппам
| Зона |
Описание |
Число SNP |
| intronic |
SNP в интронах |
213 |
exonic |
SNP в экзонах |
13 |
| UTR3 |
SNP в 3'-некодирующей области |
4 |
| UTR5 |
SNP в 5'-некодирующей области |
2 |
| ncRNA_exonic |
SNP в транскрибируемой РНК, не имеющей аннотированного кодирующего участка |
0 |
Интересно, что первые два из выбранных мной SNP попали в белок кодирующие области. Это может помочь при дальнейшей аннотации.
line147 synonymous SNV ULK4:NM_017886:exon16:c.A1536G:p.Q512Q chr3 41937051 41937051 T C het 225.009 256
line144 synonymous SNV ULK4:NM_017886:exon17:c.A1599G:p.V533V chr3 41925423 41925423 T C het 225.009 110
Аннотация по 1000 genomes
Была выполнена аннотация по базе данных 1000 genomes. Результат будет содержать частоту встречаемости найденных SNP.
Команда: perl /nfs/srv/databases/annovar/annotate_variation.pl -filter -out snp.1000g -buildver hg19 -dbtype 1000g2014oct_all snp.avinput /nfs/srv/databases/annovar/humandb/
Результаты работы программы можно найти в директории.
snp.1000g.log
snp.1000g.hg19_ALL.sites.2014_10_dropped
snp.1000g.hg19_ALL.sites.2014_10_filtered
1000g2014oct_all 0.715256 chr3 171768074 171768074 A G hom 11.3429 1
1000g2014oct_all 0.00798722 chr3 171824240 171824240 G A hom 11.3429 1
1000g2014oct_all 0.388778 chr3 171833266 171833266 G C hom 6.20226 1
1000g2014oct_all 0.460264 chr3 171861100 171861100 A G hom 5.46383 1
1000g2014oct_all 0.490016 chr3 171863825 171863825 G A hom 42.7648 2
1000g2014oct_all 0.490216 chr3 171863859 171863859 T C hom 5.46383 2
1000g2014oct_all 0.0267572 chr3 171881279 171881279 T C het 3.54557 1
1000g2014oct_all 0.246406 chr3 171886438 171886438 T G hom 9.52546 1
1000g2014oct_all 0.409545 chr3 171887000 171887000 G C het 4.13015 2
1000g2014oct_all 0.372005 chr3 171895648 171895648 T C hom 6.20226 1
1000g2014oct_all 0.683307 chr3 171900737 171900737 G A hom 11.3429 1
1000g2014oct_all 0.615216 chr3 171910642 171910642 A G hom 11.3429 1
1000g2014oct_all 0.856629 chr3 171916714 171916714 T C hom 9.52546 1
1000g2014oct_all 0.840056 chr3 171926314 171926314 C G hom 11.3429 1
1000g2014oct_all 0.377995 chr3 171926373 171926373 T C hom 7.79993 1
1000g2014oct_all 0.840455 chr3 171932935 171932935 C T hom 8.64911 1
1000g2014oct_all 0.368211 chr3 171933069 171933069 C A hom 44.7647 2
1000g2014oct_all 0.63139 chr3 171933252 171933252 G T hom 7.79993 1
1000g2014oct_all 0.654952 chr3 171949432 171949432 T C hom 11.3429 1
1000g2014oct_all 0.435903 chr3 171951720 171951720 G A hom 6.20226 1
1000g2014oct_all 0.661741 chr3 171965109 171965109 A G hom 184.999 24
1000g2014oct_all 0.0870607 chr3 171965629 171965629 A G hom 221.999 51
1000g2014oct_all 0.644768 chr3 171969077 171969077 C G hom 221.999 66
1000g2014oct_all 0.0477236 chr3 171969228 171969228 T C hom 221.999 30
1000g2014oct_all 0.0267572 chr3 171984761 171984761 C T hom 11.3429 1
1000g2014oct_all 0.156749 chr3 171985435 171985435 C T hom 11.3429 1
1000g2014oct_all 0.646366 chr3 172016000 172016000 G T hom 7.79993 1
1000g2014oct_all 0.0203674 chr3 172016753 172016753 G A hom 6.98265 2
1000g2014oct_all 0.409744 chr3 172045772 172045772 A T hom 11.3429 1
1000g2014oct_all 0.578674 chr3 172046861 172046861 T C het 225.009 95
1000g2014oct_all 0.622604 chr3 172046933 172046933 A G het 225.009 87
1000g2014oct_all 0.0654952 chr3 172055273 172055273 T C het 118.008 25
1000g2014oct_all 0.449081 chr3 172064828 172064828 A C het 149.008 35
1000g2014oct_all 0.95028 chr3 172082479 172082479 C G hom 9.52546 1
1000g2014oct_all 0.00638978 chr3 172090650 172090650 A T hom 9.52546 1
1000g2014oct_all 0.135383 chr3 172095880 172095880 C A het 54.0072 8
1000g2014oct_all 0.947284 chr3 172114474 172114474 G C hom 43.7647 2
1000g2014oct_all 0.947284 chr3 172114520 172114520 C T hom 11.3429 1
1000g2014oct_all 0.349441 chr3 172115465 172115465 T A het 161.009 48
1000g2014oct_all 0.793131 chr3 172115466 172115466 T A het 164.009 47
1000g2014oct_all 0.803914 chr3 172115640 172115640 A T het 101.008 11
1000g2014oct_all 0.00119808 chr3 172116485 172116485 C T hom 6.20226 1
1000g2014oct_all 0.277356 chr3 41291081 41291081 G A hom 221.999 25
1000g2014oct_all 0.316094 chr3 41320126 41320126 T C hom 8.64911 1
1000g2014oct_all 0.241613 chr3 41336682 41336682 T C hom 11.3429 1
1000g2014oct_all 0.140575 chr3 41349781 41349781 A T hom 9.52546 1
1000g2014oct_all 0.140775 chr3 41349977 41349977 G A hom 9.52546 1
1000g2014oct_all 0.179313 chr3 41359534 41359534 A G hom 6.98265 1
1000g2014oct_all 0.571486 chr3 41364241 41364241 G A hom 10.4247 1
1000g2014oct_all 0.990415 chr3 41371717 41371717 T C hom 38.7651 2
1000g2014oct_all 0.366214 chr3 41372090 41372090 C T hom 10.4247 1
1000g2014oct_all 0.547524 chr3 41375030 41375030 C G hom 9.52546 1
1000g2014oct_all 0.389177 chr3 41375199 41375199 G T hom 6.98265 1
1000g2014oct_all 0.393171 chr3 41375275 41375275 G C hom 7.79993 1
1000g2014oct_all 0.142572 chr3 41378582 41378582 C A hom 11.3429 1
1000g2014oct_all 0.591254 chr3 41378621 41378621 T C hom 11.3429 1
1000g2014oct_all 0.118411 chr3 41379045 41379045 C T hom 6.20226 1
1000g2014oct_all 0.102436 chr3 41379048 41379048 A T hom 6.20226 1
1000g2014oct_all 0.0944489 chr3 41398724 41398724 T C hom 11.3429 1
1000g2014oct_all 0.104633 chr3 41400196 41400196 C T hom 39.765 2
1000g2014oct_all 0.110024 chr3 41400697 41400697 T C hom 11.3429 1
1000g2014oct_all 0.0942492 chr3 41400713 41400713 C T hom 38.7651 2
1000g2014oct_all 0.0942492 chr3 41400717 41400717 T C hom 40.7649 2
1000g2014oct_all 0.0914537 chr3 41402366 41402366 T C hom 8.64911 1
1000g2014oct_all 0.109625 chr3 41402415 41402415 G A hom 38.7651 2
1000g2014oct_all 0.0984425 chr3 41404892 41404892 G A hom 9.52546 1
1000g2014oct_all 0.310503 chr3 41408203 41408203 C T het 3.01618 1
1000g2014oct_all 0.152157 chr3 41410711 41410711 C A hom 11.3429 1
1000g2014oct_all 0.106629 chr3 41412741 41412741 G C hom 10.4247 1
1000g2014oct_all 0.420128 chr3 41443267 41443267 C T het 70.0075 5
1000g2014oct_all 0.608227 chr3 41452686 41452686 A C hom 11.3429 1
1000g2014oct_all 0.00239617 chr3 41477072 41477072 T C hom 8.64911 1
1000g2014oct_all 0.586262 chr3 41478258 41478258 C A hom 11.3429 1
1000g2014oct_all 0.284345 chr3 41496752 41496752 A C hom 64.9723 3
1000g2014oct_all 0.282748 chr3 41497165 41497165 G A het 104.008 16
1000g2014oct_all 0.0133786 chr3 41498861 41498861 A C hom 11.3429 1
1000g2014oct_all 0.00239617 chr3 41502917 41502917 T C hom 7.79993 1
1000g2014oct_all 0.00858626 chr3 41504679 41504679 T C het 88.0076 8
1000g2014oct_all 0.38139 chr3 41527905 41527905 C T hom 7.79993 1
1000g2014oct_all 0.38139 chr3 41528100 41528100 G T hom 9.52546 1
1000g2014oct_all 0.996805 chr3 41528106 41528106 C T hom 11.3429 1
1000g2014oct_all 0.832468 chr3 41540052 41540052 C T hom 8.64911 1
1000g2014oct_all 0.00199681 chr3 41542282 41542282 C T hom 10.4247 1
1000g2014oct_all 0.755391 chr3 41549724 41549724 T C het 3.54366 2
1000g2014oct_all 0.804912 chr3 41554290 41554290 G A het 4.77219 1
1000g2014oct_all 0.851038 chr3 41557727 41557727 A T hom 11.3429 1
1000g2014oct_all 0.271965 chr3 41570382 41570382 C A hom 8.64911 1
1000g2014oct_all 0.46885 chr3 41589964 41589964 G C hom 7.79993 1
1000g2014oct_all 0.46885 chr3 41590054 41590054 C T hom 7.79993 1
1000g2014oct_all 0.46865 chr3 41590068 41590068 C T hom 11.3429 1
1000g2014oct_all 0.446486 chr3 41606166 41606166 C T hom 7.79993 1
1000g2014oct_all 0.434904 chr3 41606170 41606170 A G hom 9.52546 1
1000g2014oct_all 0.444888 chr3 41606251 41606251 T G hom 38.7651 2
1000g2014oct_all 0.0698882 chr3 41606466 41606466 C T hom 11.3429 1
1000g2014oct_all 0.304513 chr3 41607388 41607388 T C het 120.008 27
1000g2014oct_all 0.0700879 chr3 41607450 41607450 C T het 222.009 52
1000g2014oct_all 0.677716 chr3 41607701 41607701 C G het 137.008 36
1000g2014oct_all 0.280351 chr3 41615845 41615845 T G het 4.13164 1
1000g2014oct_all 0.319688 chr3 41615913 41615913 C T hom 6.20226 2
1000g2014oct_all 0.0445288 chr3 41635519 41635519 G C hom 11.3429 1
1000g2014oct_all 0.344649 chr3 41638984 41638984 G A hom 11.3429 1
1000g2014oct_all 0.0686901 chr3 41658278 41658278 C T hom 9.52546 1
1000g2014oct_all 0.0686901 chr3 41658297 41658297 G A hom 34.7659 2
1000g2014oct_all 0.0688898 chr3 41669123 41669123 T C hom 11.3429 1
1000g2014oct_all 0.0686901 chr3 41681725 41681725 C T het 5.46063 2
1000g2014oct_all 0.0686901 chr3 41683734 41683734 G A hom 10.4247 1
1000g2014oct_all 0.223243 chr3 41737481 41737481 T C hom 7.79993 1
1000g2014oct_all 0.224042 chr3 41737598 41737598 A G hom 11.3429 2
1000g2014oct_all 0.220847 chr3 41738596 41738596 C T hom 8.64911 1
1000g2014oct_all 0.220048 chr3 41741505 41741505 C T hom 11.3429 1
1000g2014oct_all 0.220847 chr3 41741514 41741514 A T hom 10.4247 1
1000g2014oct_all 0.158546 chr3 41748938 41748938 A C hom 11.3429 1
1000g2014oct_all 0.297524 chr3 41750323 41750323 T C hom 11.3429 1
1000g2014oct_all 0.265575 chr3 41756965 41756965 C T hom 222 38
1000g2014oct_all 0.265575 chr3 41756986 41756986 A T hom 222.085 30
1000g2014oct_all 0.31849 chr3 41759191 41759191 T C hom 220.999 34
1000g2014oct_all 0.00219649 chr3 41759457 41759457 A G het 62.0073 9
1000g2014oct_all 0.31849 chr3 41759525 41759525 C T hom 62.9723 3
1000g2014oct_all 0.1252 chr3 41779622 41779622 A T het 4.13164 1
1000g2014oct_all 0.00219649 chr3 41784422 41784422 A G hom 35.7656 2
1000g2014oct_all 0.216653 chr3 41795841 41795841 A C het 216.009 19
1000g2014oct_all 0.302316 chr3 41803385 41803385 C T hom 9.52546 1
1000g2014oct_all 0.305312 chr3 41813104 41813104 T C hom 43.7647 2
1000g2014oct_all 0.290535 chr3 41813108 41813108 C T hom 39.765 2
1000g2014oct_all 0.291334 chr3 41824696 41824696 - T het 4.4191 1
1000g2014oct_all 0.0814696 chr3 41831203 41831203 C T het 206.009 30
1000g2014oct_all 0.0830671 chr3 41836919 41836919 G A hom 7.79993 1
1000g2014oct_all 0.0830671 chr3 41836920 41836920 T C hom 10.4247 1
1000g2014oct_all 0.870008 chr3 41841716 41841716 A C hom 221.999 31
1000g2014oct_all 0.0830671 chr3 41841873 41841873 C T het 78.0075 12
1000g2014oct_all 0.0830671 chr3 41843391 41843391 A G hom 7.79993 1
1000g2014oct_all 0.0830671 chr3 41843443 41843443 A G hom 11.3429 1
1000g2014oct_all 0.0828674 chr3 41850685 41850685 A C het 3.54557 1
1000g2014oct_all 0.0788738 chr3 41860955 41860955 A G het 171.009 25
1000g2014oct_all 0.253594 chr3 41863817 41863817 A G hom 9.52546 1
1000g2014oct_all 0.169129 chr3 41863859 41863859 G A hom 11.3429 1
1000g2014oct_all 0.313099 chr3 41864697 41864697 A T het 6.20096 2
1000g2014oct_all 0.313099 chr3 41864872 41864872 C T hom 11.3429 1
1000g2014oct_all 0.171925 chr3 41869847 41869847 A T hom 6.98265 1
1000g2014oct_all 0.14976 chr3 41872527 41872527 G T hom 7.79993 1
1000g2014oct_all 0.172724 chr3 41877414 41877414 T C het 205.009 43
1000g2014oct_all 0.315096 chr3 41882323 41882323 A G hom 6.20226 1
1000g2014oct_all 0.315096 chr3 41882357 41882357 T C het 4.13164 1
1000g2014oct_all 0.0828674 chr3 41886884 41886884 A C hom 11.3429 1
1000g2014oct_all 0.142372 chr3 41888671 41888671 G C het 4.77219 1
1000g2014oct_all 0.0828674 chr3 41900656 41900656 A G het 62.0073 10
1000g2014oct_all 0.140176 chr3 41900774 41900774 T C het 181.009 16
1000g2014oct_all 0.315296 chr3 41900951 41900951 C A hom 221.999 24
1000g2014oct_all 0.000599042 chr3 41901030 41901030 C A het 225.009 32
1000g2014oct_all 0.311901 chr3 41902964 41902964 C T hom 11.3429 1
1000g2014oct_all 0.311901 chr3 41902972 41902972 G C hom 11.3429 1
1000g2014oct_all 0.169529 chr3 41903491 41903491 A T hom 11.3429 1
1000g2014oct_all 0.142372 chr3 41903601 41903601 G T hom 10.4247 1
1000g2014oct_all 0.0828674 chr3 41904576 41904576 A T hom 7.79993 1
1000g2014oct_all 0.167332 chr3 41909219 41909219 C T het 4.13164 1
1000g2014oct_all 0.851238 chr3 41910571 41910571 T C hom 10.4247 1
1000g2014oct_all 0.832468 chr3 41923872 41923872 T C het 106.008 12
1000g2014oct_all 0.919329 chr3 41925423 41925423 T C het 225.009 110
1000g2014oct_all 0.0840655 chr3 41936827 41936827 G A het 147.008 91
1000g2014oct_all 0.00279553 chr3 41937051 41937051 T C het 225.009 256
1000g2014oct_all 0.91893 chr3 41938500 41938500 G C het 168.009 21
1000g2014oct_all 0.086262 chr3 41939781 41939781 G A het 167.009 37
1000g2014oct_all 0.698083 chr3 41939993 41939993 T A het 225.009 67
1000g2014oct_all 0.0840655 chr3 41949301 41949301 T C het 134.008 16
1000g2014oct_all 0.940495 chr3 41952781 41952781 C T het 225.009 95
1000g2014oct_all 0.0840655 chr3 41952852 41952852 T C het 225.009 76
1000g2014oct_all 0.911142 chr3 41973062 41973062 T C het 5.46063 2
1000g2014oct_all 0.0838658 chr3 41977464 41977464 A C het 225.009 51
1000g2014oct_all 0.0836661 chr3 41978738 41978738 A T het 194.009 29
1000g2014oct_all 0.910942 chr3 41983615 41983615 G C het 5.46063 2
1000g2014oct_all 0.915535 chr3 41996275 41996275 A G het 225.009 47
1000g2014oct_all 0.0966454 chr3 41996304 41996304 G A het 217.009 36
1000g2014oct_all 0.490415 chr3 52720080 52720080 A C hom 221.999 74
1000g2014oct_all 0.125799 chr3 52720489 52720489 T A hom 11.3429 1
1000g2014oct_all 0.489018 chr3 52722335 52722335 A C het 100.062 8
Были посчитаны частоты встречаемости SNP. Максимальная составила 0.996805, а минимальная - 0.000599042.
Аннотация по Gwas
Была выполнена аннотация по базе данных Gwas. Она позволяет понять, с чем, из ранее описанного в литературе, ассоциированы SNP.
Команда: perl /nfs/srv/databases/annovar/annotate_variation.pl -regionanno -out snp.gwas -build hg19 -dbtype gwasCatalog snp.avinput /nfs/srv/databases/annovar/humandb/
Результаты работы программы можно найти в директории. Было найдено четыре SNP, ассоциированных с уровнем кровяного давления, уровнем адипонектина и ростом человека.
Использую поиск по GWAS Catalog, можно подробнее изучить все исследования, ассоциации и испытания, связанные с конкретным SNP.
Поиск по базе осуществлялся с помощью rs идентификатора, полученного из базы данных
snp.gwas.hg19_gwasCatalog
snp.gwas.log
gwasCatalog Name=Blood pressure chr3 41877414 41877414 T C het 205.009 43
gwasCatalog Name=Adiponectin levels chr3 52720080 52720080 A C hom 221.999 74
gwasCatalog Name=Height chr3 171926373 171926373 T C hom 7.79993 1
gwasCatalog Name=Height chr3 171969077 171969077 C G hom 221.999 66
Аннотация по Clinvar
Была выполнена аннотация по базе данных Clinvar.
Команда: perl /nfs/srv/databases/annovar/annotate_variation.pl -filter -out snp.clin -dbtype clinvar_20150629 -buildver hg19 snp.avinput /nfs/srv/databases/annovar/humandb/
Результаты работы программы можно найти в директории.
snp.clin.hg19_clinvar_20150629_dropped
snp.clin.hg19_clinvar_20150629_filtered
snp.clin.log
Оказалось, что в Clinvar не аннотровано ни одного SNP. Как говорится, отрицательный результат - тоже результат.
Ознакомиться с полной информацией о найденных SNP вы можете в таблице.
Положение ридов на выравнивании вы можете увидеть на рисунке 18.
Рисунок 18. Распределение ридов.
|
Крайняя таблица. Использовавшиеся команды.
| Команда |
Результат |
| fastqc chr3.fastq |
Анализ качества ридов |
| java -jar /usr/share/java/trimmomatic.jar SE -phred33 chr3.fastq chr3_outfile.fastq TRAILING:20 MINLEN:50 |
Очистка чтений |
| export PATH=${PATH}:/home/students/y06/anastaisha_w/hisat2-2.0.5 |
Прописывает временный путь до исполняемой программы |
| hisat2-build chr3.fasta chr3 |
Индексирует референсную последовательность, записывает результат в *.ht2 файлы |
| hisat2 -x chr3 -U chr3_outfile.fastq --no-spliced-alignment --no-softclip > alignment.sam |
Строит выранивание прочтения и референса, записывает результат в .sam файл |
| samtools view alignment.sam -b -o alignment.bam |
Переводит выравнивание в бинарный формат |
| samtools sort alignment.bam -T out_sort.txt -o alignment_sorted.bam |
Индексирует референсную последовательность, записывает результат в *.ht2 файлы |
| samtools index alignment_sorted.bam |
Индексирует отсортированный .bam файл |
| samtools depth alignment_sorted.bam >depth_sorted.tsv |
Создаёт файл, содержащий информацию о глубине прочтения каждого SNP |
| perl /nfs/srv/databases/annovar/convert2annovar.pl -format vcf4 /nfs/srv/databases/ngs/pavel-kravchenko/snp.vcf > /nfs/srv/databases/ngs/pavel-kravchenko/snp.avinput |
Подготовка файла для аннотирования |
| /nfs/srv/databases/annovar/annotate_variation.pl -filter -out snp.rs -build hg19 -dbtype snp138 snp.avinput /nfs/srv/databases/annovar/humandb/ |
Поиск rc SNP по базе данных dbSNP |
| perl /nfs/srv/databases/annovar/annotate_variation.pl -out snp.refgene -build hg19 snp.avinput /nfs/srv/databases/annovar/humandb/ |
Аннотация по базе данных RefGene |
| perl /nfs/srv/databases/annovar/annotate_variation.pl -filter -out snp.1000g -buildver hg19 -dbtype 1000g2014oct_all snp.avinput /nfs/srv/databases/annovar/humandb/ |
Аннотация по базе данных 1000 genomes |
| perl /nfs/srv/databases/annovar/annotate_variation.pl -regionanno -out snp.gwas -build hg19 -dbtype gwasCatalog snp.avinput /nfs/srv/databases/annovar/humandb/ |
Аннотация по базе данных Gwas |
| perl /nfs/srv/databases/annovar/annotate_variation.pl -filter -out snp.clin -dbtype clinvar_20150629 -buildver hg19 snp.avinput /nfs/srv/databases/annovar/humandb/ |
Аннотация по базе данных Clinvar |
Ссылки
- Распределение файлов с ридами
- FastQC
- Trimmomatic
- Руководство Trimmomatic
- samtools
- bcftools
- annotate_variation.pl
- convert2annovar.pl.
- Adiponectin